Spinal Muscular Atrophy
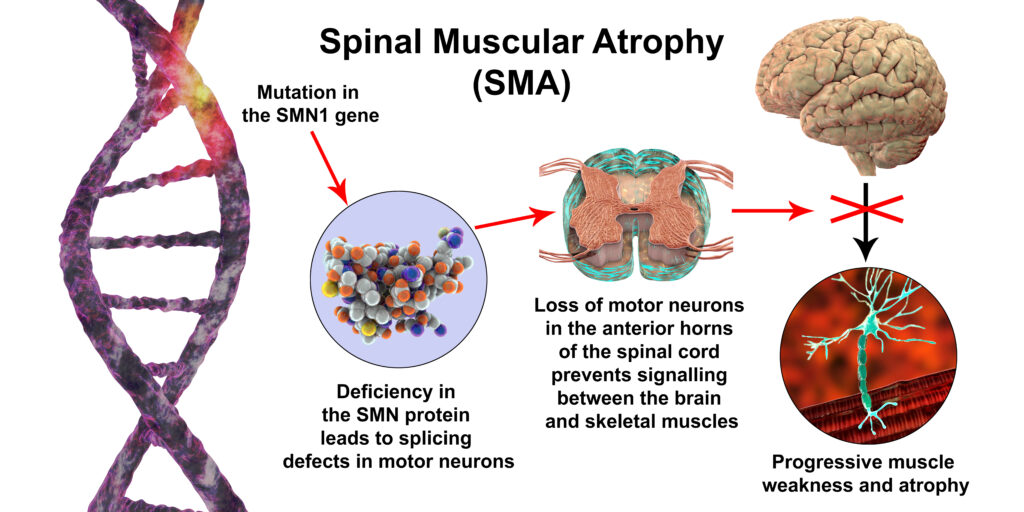
Spinal Muscular Atrophy, more commonly known as SMA, is a group of genetic diseases that affects both the central nervous system (CNS) which is the brain and spinal cord, and the peripheral nervous system (PNS) which includes all the other nerves in the body. The disease typically progresses with increasing muscle weakness as the child ages. Weakness and atrophy (muscle wasting) are more pronounced toward the center of the body as opposed to the extremities. SMA is usually symmetrical on both sides of the body.
Specifically affected in SMA are motor nerves in the spinal cord in the anterior horn (the gray matter next to the central canal). This interrupts messages sent from the brain to the body. The message for movement cannot be fulfilled by the use of skeletal muscle to voluntarily create movement. As the body’s muscles do not receive messages from the nerves, muscles atrophy and shrink over time. Particularly affected is the movement of the face, tongue, throat, arms, chest, and legs. This affects functions such as breathing, swallowing, speaking, and walking.
Body functions that are not affected by SMA include mental functions such as learning and social skills. Sensory nerves are not affected therefore intact sensation or feeling in the body is maintained. Vision and hearing are not affected. Bowel and bladder function are unaffected.
Genetics
SMA is most commonly caused by a mutation or missing gene, which makes it a genetic (inherited) disease. SMN is a protein in the body that is necessary for survival as a deficiency will result in selective motor neuron loss. The affected gene causing SMA is named the survival motor neuron gene 1(SMN1) which is located on chromosome 5q. This gene is responsible for maintaining the health and function of motor neurons, which are the nerves that cause movement in the body. The gene SMN2 gives the instruction to the body to make the SMN1 gene. If SMN1 is absent and SMN2 is lacking, SMN cannot be produced. Individuals who lack the SMN1 gene have SMA. although some individuals without SMN1 may rely on SMN2 to produce SMN.
To have SMA, individuals need to inherit a recessive gene (non-dominant) from both parents. Inheriting only one recessive gene from one parent will not lead to the disease, but the individual will be a carrier of SMA. There have been rare cases where the genetic factors are not clear.
There are many questions about life expectancy with SMA. Traditionally, the lower type of SMA has a lower life expectancy. The highest type IV has a life expectancy that is the same as the general population. However, with the development of new treatments to improve SMA, life expectancies are improving. Also, researchers are constantly increasing options to improve breathing, mobility, and other complications of SMA.
Types of SMA
There are five types of SMA that are based on best motor function. A classification system for SMA was put forth in 1991 by the Muscular Dystrophy Association (MDA). The degree of severity varies within types. Types can be across categories. Only about 25% of individuals with SMA are able to be clearly classified into just one type. You may hear this ‘Type’ classification system used.
Original SMA Classification System
| SMA Type | Age of Diagnosis/Onset | Clinical signs | Ability |
| Type 0 SMA | Prenatal | · Decreased fetal movement· Poor or lack of reflexes
· Facial paralysis · Heart septum defect · Joint contractures and skeletal issues · Muscle twitching · Difficulty swallowing and feeding · Respiratory failure |
Support for breathing is required |
| Type I SMAAKA Werdnig-Hoffmann disease | 0-6 months | · Low body tone (frog leg posture)· Poor head control
· Low or non-responsive reflexes · Belly breathing from poor strength in intercostal muscles (those between the ribs), but functioning diaphragm · Aspiration risk due to oral motor weakness · Unaffected mentation |
Unable to roll or sit independently |
| Type II SMA | Less than 18 months | · Typically, progressive weakness in the legs but not as much in the arms· Low tone, without reflexes
· Bone complications due to weak muscles such as scoliosis contractures, stiffening of the jaw · Restrictive lung disease · Unaffected mentation |
Ability to sit but not stand/walk |
| Type III SMAAKA Kugelberg-Welander disease | Greater than 18 months,Subtypes:
3a 18 mos.-3 years 3b >3 years |
· Leg weakness greater than arms· Little to no respiratory muscle weakness or scoliosis
· Difficulty running, climbing stairs, attempting to stand · Unaffected mentation · Unaffected life expectancy |
Walk but may be delayed |
| Type IV SMA | Greater than 21 years or in adulthood | · 5% of cases· Onset in adulthood, most often around age 30 but can be earlier or later
· Mild to moderate leg weakness · Unaffected mentation · Unaffected life expectancy |
Walk independently or with mobility assistance devices |
| SMA not linked to chromosome 5 (SMN1 or SMN protein) | Varies | · Varies in severity· May include muscles further away from the center of the body | Varies |
Current SMA Classification System
SMA is a health issue that changes over the course of time. Some individuals will be classified as above whereas others will move from one type to another based on functional ability. The shift can be in gains or losses of function depending on the individual. Therefore, a more practical classification system was developed to indicate the current level of function rather than function at the time of diagnosis. This classification system is sectioned into three categories with subcategories being added as more research is conducted. The classifications are Nonsitters, Sitters, and Walkers. No one fits perfectly into any one category and changes in category are frequent.
| Category | Type Classification | Characteristics |
| Nonsitters | Mostly Type I | · Breathing and feeding difficulties· Bell-shape thorax deformity
· Fasciculations of the tongue · Absent hip flexion, head lag |
| Sitters | Mostly Type II & III | · Achieve sitting but not walking· Muscle weakness is more profound in the legs than arms
· Development of joint contractures, jaw ankylosis, frequently scoliosis · Most do not have breathing swallowing issues, however, intercostal muscle weakness can lead to restrictive lung disease and respiratory insufficiency · SMA may progress between ages 5 and 15, especially at puberty |
| Walkers | Type II or IV | · Those who achieve or maintain walking ability· Less leg function than arm weakness
· Typically, no breathing or swallowing difficulty, however, over a lifetime, walking and breathing may very slowly and slightly decrease · 95% of early sitters may walk by 18 months · 50% of late sitters may walk by 18 months · Life expectancy is equal to the general population |
| Adult Onset | Type IV | · Symptoms may appear as discomfort or pain in leg muscles· Legs may become weak requiring splinting or braces to maintain walking
· Swallowing and breathing are typically not affected |
Symptoms/Diagnosis
Muscle weakness is generally the first symptom of SMA. If muscle weakness is severe, especially with more weakness toward the center of the body and less in the extremities, the diagnosis will be fairly clear. Mild weaknesses may not lead to the SMA diagnosis directly.
Genetic testing using a blood sample to assess for mutations or deletions in the SMN1 and SMN2 genes is the definitive test. It will also be able to detect someone who is a carrier for SMA. If a biological male and biological female are both carriers, they may choose to make a personal decision to use a donor to reduce the risk of the disease in a child. The genetic test for SMA is 95% reliable.
Electromyography (EMG) or testing of muscle function by recording the electrical activity of the muscles can be performed by comparing muscle contraction and relaxation.
Nerve conduction studies (NCS) may be performed at the time of the EMG. This test assesses the ability of a nerve to transmit messages.
![]()
This graphic was reprinted with permission from SMA News Today.
Muscle biopsy may be assessed where an extremely small fragment of a muscle is removed from the body and microscopically examined for changes indicating neuromuscular disease.
Biomarkers Changes in certain features in the body or blood known as biomarkers can indicate diseases. SMN2 copy number inversely correlates with a diagnosis of SMA and an indication of severity. EMG measures of compound muscle action potentials, motor unit estimation, and electrical impedance myography, are predictive of SMA and medication estimations for treatment.
Other blood tests including SMN mRNA and protein levels can reveal waste materials from muscle destruction.
Treatment
Medications are available for specific types of SMA.
Nusinersen (SPINRAZA) increases the production of the SMN protein through modulation of SMN2 splicing which maintains motor neurons. This medication is delivered intrathecally (in the space between the spinal cord and the membrane that contains it) via a spinal injection. Available for children and adults with any type of SMA.
Onasemnogene abeparvovec-xioi (Zolgensma) is an SMN1 gene replacement therapy. SMN protein production is improved by the transfer of the SMN1 gene entering neural (nerve) cells using adeno-associated virus stereotypes. These viruses are non-pathogenic (will not make your child sick.) They pass easily into cells, which are commonly used in gene therapy because of their safety. More information is in the Pillay, 2017 article in the reference section. It is available for children less than two years old with infantile-onset SMA. The goal is to improve muscle movement, function, and survival.
Risdiplam (Evrysdi) is an oral medication to stimulate SMN3 gene expression of SMN protein. It is for children two months and older.
The number of studies of SMA treatments has been bountiful since the discovery of the SMN gene. This has led to laboratory studies as well as clinical (with humans) studies that are ongoing. These studies include small molecule therapy to increase SMN protein, RNA-based therapy to increase SMN2 into the RNA molecule for increased SMN2 expression, and gene therapy to replace the missing SMN1 gene.
More studies are rapidly being performed. Be sure to check the website: https://clinicaltrials.gov/ for new clinical trials sponsored by the U.S. government. Information about individual clinical trials for SMA will provide you with knowledge about the thoughts of researchers and the latest trial information. It is important to know what will be coming so you can prepare and make well-informed decisions for the health of your child.
Rehabilitation Therapies
A team of healthcare professionals will be caring for your child. Each brings their specialty to meet individual needs as they work in unison.
Diagnosis is made by a geneticist or a neurologist who is consulted if SMA is suspected. This can be an emotionally draining experience. Having information about the disease is helpful in understanding what medical treatment options will be available for your child as well as how to maintain their quality of life and experiences. This person will contribute to the Individualized Education Program or Plan (IEP) that will be needed in school to meet the specific needs of your child. They can also assist with further family planning. They will be able to explain options for research therapies if you are interested in pursuing this treatment pathway.
Mental wellness can be enhanced by a psychologist who works with children and families. This individual is helpful in navigating the emotions and effects on your child, your family, and you. This person will also contribute to the Individualized Education Program (IEP) that will be needed in school to meet the specific needs of your child.
Specialty healthcare will be provided by a specialist in pediatric neuromuscular disorders, a pediatric physiatrist, or an adult physiatrist (based on age). It is the medical doctor who will care for the functional needs of your child both in promoting habilitation (learning new skills) and rehabilitation (relearning skills). This individual can lead the healthcare team in organizing and providing care including medical treatment, equipment, therapy, medications, and testing. They will be aware of research and new treatments that will be beneficial.
Schooling Educators such as the school principal, teachers, and educational aides may be required to provide support for your child’s learning. Involving school personnel through the use of the IEP is essential. With SMA, learning ability is not affected, but the motor requirements for breathing and mobility equipment, note-taking, physical activity, feeding assistance, rest time, and other issues may require adaptive equipment. The IEP is a plan created and documented concerning the needs of your child and how the school system will meet them.
Equipment for maximum independence will be recommended by physical, occupational, and speech/language therapists. Positioning equipment is needed to maintain body alignment which supports breathing, the skeletal system, and digestion, among many other issues. Equipment may include sleep systems, seating, wheelchairs, walkers, braces, and bathing equipment.
Therapies for spinal muscle atrophy are based on individual needs. Not every individual will need all these treatments, however, knowing that they are available is helpful to improve health as well as to avoid health issues.
Breathing A pediatric or adult pulmonologist and respiratory therapist will prescribe and coordinate respiratory treatments as needed. This may include mechanical ventilation which may require a tracheostomy, non-invasive mechanical ventilation that does not require a tracheostomy, suction, insufflation, nebulizer treatments, and breathing exercises such as incentive spirometry. Respiratory infections can occur more easily therefore practicing hand washing, breathing exercises, safe distancing, and isolation for individuals who are ill is important.
Some children may have sleep apnea when at rest. Be sure to have an evaluation and monitor for symptoms such as snoring, snorting, periods of not breathing, or morning headache when lying. Breathing issues can lead to other health conditions such as affecting heart function.
Oral Motor Skills can be strengthened and improved through the assistance of a speech/language pathologist to help with chewing, swallowing, and improvements in speech articulation. If swallowing is an issue, alternative feeding methods may be used such as tube feedings.
Communication Oral motor issues can interrupt communication. Devices that will help with communication are available that require slight hand movement or use eye-gaze technology.
Nutrition Eating concerns of chewing, swallowing, and physically bringing food to the mouth may be issues. It is necessary to ensure that proper nutrition is provided. It may be difficult to maintain caloric intake for bone health, growth, and development or to avoid overfeeding. Height, weight, and body mass index (BMI) should be continuously monitored. A dietitian can assist with providing an appropriate diet that works best for your child. Use of adaptive equipment may be provided by an occupational therapist.
Gastrointestinal (GI) Issues Challenges with chewing, liquid diets, and reduced movement can slow the digestive system. This can result in acid reflux, feeling full due to slow stomach emptying or bowel function, vomiting, and constipation. Issues with metabolism, high blood pressure, blood sugar, and breaking down fat can be challenges. Exercise improves digestion and sugar metabolism. Standing therapy also assists digestion. Gastroenterologists specialize in GI issues. Medications may be needed for the above issues.
Orthopedic: Issues of the skeleton are a concern with SMA. An orthopedist, a healthcare professional who is educated in the care of the bones, is helpful in not only treating bone issues but also avoiding problems.
Stretching the joints of the body is important to keeping them supple. It is an activity that may be provided to your child if they cannot move their body independently. This helps avoid contractures, and cramping as well as provides general comfort to the body.
Splints and Braces may be used to hold the body in position, stabilize joints, and promote movement. These may be used on the arms, legs, torso, neck, or wherever positioning is needed. Some are worn only at night.
Contractures or stiffening of the joints is a common issue in SMA as movement is challenged. Therapeutic treatment will be provided and taught to the child and family members to maintain or improve flexibility and slow the progression of muscle wasting as well as providing movement and exercise. Contractures may be reduced through orthopedic surgery or serial casting where the joint is released over time.
Positioning is important to the maintenance and function of the body. Equipment to place the body in anatomical alignment will assist with keeping the body in effective, functional form. Stretching exercises, splints, and serial casting may be helpful.
Standing frames assist with assuming an upright position which allows the child to see the world at their natural height, improves personal interactions in some situations, improves bone development, and can aid with posture and digestion. Be sure to check with your child’s healthcare professional if they are physically ready for this activity to avoid unexpected complications, especially to the bones or vascular systems.
Scoliosis (spine shapes like an ‘S’) and kyphosis (spine bends forward like a ‘C’) are concerns because of weakness in the muscles of the trunk. Muscular imbalances can pull the vertebrae of the spine out of alignment and reduce the ability to breathe effectively. Management of scoliosis can be achieved through exercise, bracing, and if necessary, surgical intervention. If surgical intervention is required, you may have the choice between rigid stabilization of the spine or flexible stabilization. The newer flexible stabilization rods allow some slight trunk movement once healed.
Hip alignment is a critical element when in bed as well as when sitting. Being sure there is alignment in the hips as well as stretching of hip muscles, will keep the body in a good position which will avoid issues of hip contractures, misalignment, and scoliosis.
Mobility and Exercise are critical for everyone. The body craves movement. Exercise benefits breathing, digestion, and mental and general health. Mobility devices such as wheelchairs, walkers, splints, and braces will assist with your child’s movement through their environment. This creates independence in their world when they are developmentally ready to do so. Exercise through therapy will be prescribed on an individual basis. Other options for exercise might include movement gaming, aquatic therapy, adaptive sports, or anything that encourages body functioning.
Bone Health When body weight is not put through the bones, minerals tend to decrease making the bones brittle which can result in an easily fractured bone. Osteopenia is a loss of bone density which leads to osteoporosis. Bone density testing should be performed annually with individuals who have SMA of all ages to catch bone density early so treatment can begin. Exercise and standing in a standing frame helps maintain bone density but some cases are more severe and require medication.
The Palliative Care team consists of a group of individuals, including specially educated nurses, who will assist with medical support for quality of life needs. This service is not just for end-of-life care but will ensure your child’s and your needs are met with support. SMA can evolve in many directions. This group may enter into care with your child or move out of care however needs change.
History
1891 SMA was first described in the medical literature when the condition was noted in two infant brothers by Gundo Werdnig. Further descriptions of progressive weakness in infants were described in 1893-1900 by Johan Hoffmann when the term ‘spinal muscle atrophy’ was given. Werdnig-Hoffmann became the name for Type I SMA however, their descriptions were of intermediate cases.
Severe cases were presented in 1899 and 1903. Milder cases of walking and standing with SMA were presented in the 1950s. There was controversy about the types and classifications until 1991 when the Muscular Dystrophy Association sponsored the classification system at an International Consortium on Spinal Muscular Atrophy based on three SMA types and age of onset.
It was not until 1995 that SMA was determined to be caused by a deletion in the SMN1 gene on chromosome 5Q13 and SMN2 by the Melki laboratory. This was a pivotal discovery as prior to this discovery, there was no specific reason known for the development of SMA, therefore developing a treatment was undeterminable. Within five years of the genetic discovery, animal models could be created to study SMA and begin to develop treatments for it.
In 2007, Wang and colleagues developed the first clinical guideline for the care of individuals with SMA including aging with SMA.
Since the understanding of the genetic issues, The National Institute of Neurological Disorders and Stroke, foundations, and pharmacologists and biotechnologists have been developing therapies to increase or improve SMN 1 and SMN2 using small molecule therapy, RNA therapy, and gene therapy. The results have been significant with the following therapeutic treatments available:
- 2016 Nusinersen (SPINRAZA) was approved by the FDA for all types of SMA.
- 2017 Onasemnogene abeparvovec-xioi (Zolgensma) was approved by the FDA focusing on SMN1 gene replacement.
- 2020 Risdiplam (Evrysdi) was approved by the FDA to stimulate SMN2 gene expression of the SMN protein. This is the first oral medication for SMA treatment.
Research has steadily increased with more studies being conducted and in various states of clinical trials. Also, it is important to remember that many other researchers have helped to improve the lives of individuals with SMA due to their research in the improvements in breathing, medications, treatment of scoliosis, and other bone issues, as well as a focus on quality of life issues among many others.
Facts and Figures
SMA is the most common cause of death due to genetic alteration in infants.
One in every 6,000 babies is born with SMA. One in 11,000 individuals of all ages are affected by SMA.
Those who carry the genetic anomaly of SMA but do not have the condition are 1 in 54.
The most common type of SMA is Werdnig-Hoffmann’s disease which is responsible for 80% of cases. Kugelberg-Welander disease is another type that is diagnosed in early childhood.
Development of SMA in infancy and early childhood is associated with poorer outcomes than if it develops later in life.
Resources
If you are looking for more information on spinal muscular atrophy or have a specific question, our Information Specialists are available business weekdays, Monday through Friday, toll-free at 800-539-7309 from 7:00 am to 12:00 am midnight ET.
Additionally, the Reeve Foundation maintains a SMA fact sheet with additional resources from trusted Reeve Foundation sources. Check out our repository of fact sheets on hundreds of topics ranging from state resources to secondary complications of paralysis.
We encourage you to reach out to spinal muscular atrophy support groups and organizations, including:
Cure SMA provides support programs for people living with SMA and their families, as well as funding and directing comprehensive research for treatments and a cure. Toll-free 1-800—886-1762. https://www.curesma.org/
Muscular Dystrophy Association (MDA) provides services and supports research for a group of hereditary muscle-destroying disorders, including spinal muscular atrophies. Toll-free 1-800-572-1717. Search under “Diseases” at https://www.mda.org/disease/spinal-muscular-atrophy
National Institute of Neurological Disorders and Stroke (NINDS) provides information on SMA. https://www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy
SMA Foundation hopes to accelerate the development of a treatment or cure for SMA. Toll-free 1-877-FUND-SMA. https://smafoundation.org/about-sma/sma-organizations-worldwide/
Support Groups for SMA provides a list of SMA support groups. https://spinalmuscularatrophy.net/support-groups
A clinical practice guideline for SMA is available at:
A Guide to the 2017 International Standards of Care for SMA, TREAT-NMD, Neuromuscular Network: https://smacare.guide/
References
Belter L, Jarecki J, Reyna SP, Cruz R, Jones CC, Schroth M, O’Toole CM, O’Brien S, Hall SA, Johnson NB, Paradis AD. The Cure SMA Membership Surveys: Highlights of Key Demographic and Clinical Characteristics of Individuals with Spinal Muscular Atrophy. J Neuromuscul Dis. 2021;8(1):109-123. doi: 10.3233/JND-200563. PMID: 33104036; PMCID: PMC7902958. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7902958/pdf/jnd-8-jnd200563.pdf
Belter L, Mazzella A, O’Brien S, Jarecki J. Knowledge of Genetic Test Results Among Caregivers and Individuals with Spinal Muscular Atrophy. PLoS One. 2022;17(11):e0276756. Published 2022 Nov 8. doi:10.1371/journal.pone.0276756. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9642888/pdf/pone.0276756.pdf
Belter L, Cruz R, Kulas S, McGinnis E, Dabbous O, Jarecki J. Economic Burden of Spinal Muscular Atrophy: An Analysis of Claims Data. J Mark Access Health Policy. 2020;8(1):1843277. Published 2020 Nov 8. doi:10.1080/20016689.2020.1843277
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7655070/
Chiriboga CA. Nusinersen for the Treatment of Spinal Muscular Atrophy. Expert Rev Neurother. 2017 Oct;17(10):955-962. doi: 10.1080/14737175.2017.1364159. Epub 2017 Sep 8. PMID: 28884620.
https://pubmed.ncbi.nlm.nih.gov/28884620/
Darras BT, Farrar MA, Mercuri E, Finkel RS, Foster R, Hughes SG, Bhan I, Farwell W, Gheuens S. An Integrated Safety Analysis of Infants and Children with Symptomatic Spinal Muscular Atrophy (SMA) Treated with Nusinersen in Seven Clinical Trials. CNS Drugs. 2019 Sep;33(9):919-932. doi: 10.1007/s40263-019-00656-w. PMID: 31420846; PMCID: PMC6776494. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6776494/
Jablonka S, Hennlein L, Sendtner M. Therapy Development for Spinal Muscular Atrophy: Perspectives for Muscular Dystrophies and Neurodegenerative Disorders. Neurol Res Pract. 2022;4(1):2. Published 2022 Jan 4. doi:10.1186/s42466-021-00162-9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8725368/pdf/42466_2021_Article_162.pdf
Jones CC, Cook SF, Jarecki J, et al. Spinal Muscular Atrophy (SMA) Subtype Concordance in Siblings: Findings from the Cure SMA Cohort. J Neuromuscul Dis. 2020;7(1):33-40. doi:10.3233/JND-190399.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7029365/pdf/jnd-7-jnd190399.pdf
Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin. 2015 Nov;33(4):831-46. doi: 10.1016/j.ncl.2015.07.004. PMID: 26515624; PMCID: PMC4628728. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4628728/
Kolb SJ, Kissel JT. Spinal Muscular Atrophy: A Timely Review. Arch Neurol. 2011 Aug;68(8):979-84. doi: 10.1001/archneurol.2011.74. Epub 2011 Apr 11. PMID: 21482919; PMCID: PMC3860273. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3860273/pdf/nihms518569.pdf
Mazzella A, Curry M, Belter L, Cruz R, Jarecki J. “I Have SMA, SMA Doesn’t Have Me”: A Qualitative Snapshot into the Challenges, Successes, and Quality of Life of Adolescents and Young Adults with SMA. Orphanet J Rare Dis. 2021;16(1):96. Published 2021 Feb 22. doi:10.1186/s13023-021-01701-y. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7898731/pdf/13023_2021_Article_1701.pdf
Ogbonmide T, Rathore R, Rangrej SB, Hutchinson S, Lewis M, Ojilere S, Carvalho V, Kelly I. Gene Therapy for Spinal Muscular Atrophy (SMA): A Review of Current Challenges and Safety Considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus. 2023 Mar 15;15(3):e36197. doi: 10.7759/cureus.36197. PMID: 37065340; PMCID: PMC10104684. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10104684/
Ojala KS, Reedich EJ, DiDonato CJ, Meriney SD. In Search of a Cure: The Development of Therapeutics to Alter the Progression of Spinal Muscular Atrophy. Brain Sci. 2021;11(2):194. Published 2021 Feb 5. doi:10.3390/brainsci11020194. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7915832/pdf/brainsci-11-00194.pdf
Paik J. Risdiplam: A Review in Spinal Muscular Atrophy. CNS Drugs. 2022 Apr;36(4):401-410. doi: 10.1007/s40263-022-00910-8. Epub 2022 Mar 13. PMID: 35284988. https://link.springer.com/article/10.1007/s40263-022-00910-8
Peterson I, Cruz R, Sarr F, Stanley AM, Jarecki J. The SMA Clinical Trial Readiness Program: Creation and Evaluation of a Program to Enhance SMA Trial Readiness in the United States. Orphanet J Rare Dis. 2020;15(1):118. Published 2020 May 22. doi:10.1186/s13023-020-01387-8. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7564894/pdf/13023_2020_Article_1387.pdf
Pillay S, Zou W, Cheng F, Puschnik AS, Meyer NL, Ganaie SS, Deng X, Wosen JE, Davulcu O, Yan Z, Engelhardt JF, Brown KE, Chapman MS, Qiu J, Carette JE. Adeno-Associated Virus (AAV) Serotypes Have Distinctive Interactions with Domains of the Cellular AAV Receptor. J Virol. 2017 Aug 24;91(18):e00391-17. doi: 10.1128/JVI.00391-17. PMID: 28679762; PMCID: PMC5571256. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5571256/pdf/e00391-17.pdf
Ross, LF, Kwon JM. Spinal Muscular Atrophy: Past, Present, and Future. NeoReviews, August 7, 2019. https://web.archive.org/web/20200228122152id_/https://pdfs.semanticscholar.org/f1ee/2031cada7d7d3e77ea248ba09863e5fd05d4.pdf
Wirth B. An Update of the Mutation Spectrum of the Survival Motor Neuron Gene (SMN1) in Autosomal Recessive Spinal Muscular Atrophy (SMA). Hum Mutat. 2000;15(3):228-37. doi: 10.1002/(SICI)1098-1004(200003)15:3<228:AID-HUMU3>3.0.CO;2-9. PMID: 10679938.
https://pubmed.ncbi.nlm.nih.gov/10679938/
Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annual Review of Genomics and Human Genetics 2020 21:1, 231-261. https://www.annualreviews.org/doi/pdf/10.1146/annurev-genom-102319-103602
