Atrofia muscular espinal
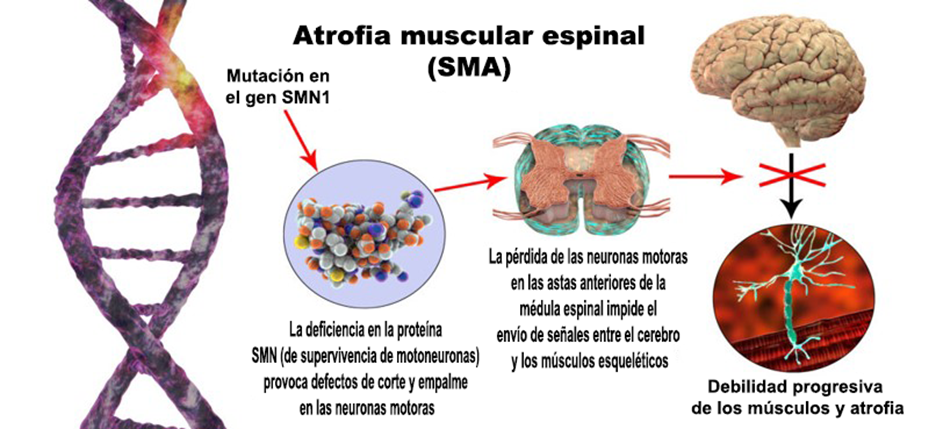
La atrofia muscular espinal, mejor conocida como SMA, es un grupo de enfermedades genéticas que afectan el sistema nervioso central (CNS, por sus siglas en inglés) que consiste en el cerebro, en la médula espinal y en el sistema nervioso periférico (PNS, por sus siglas en inglés), que incluye a todos los demás nervios del cuerpo. La enfermedad normalmente avanza con debilidad muscular en aumento a medida que el menor cumple años. La debilidad y la atrofia (desgaste muscular) son más pronunciadas en el centro del cuerpo y no en las extremidades. La SMA normalmente es simétrica en ambos lados del cuerpo.
Los nervios de la médula espinal en el asta anterior (la materia gris junto al canal central) son los que se ven específicamente afectados. Esto interrumpe los mensajes que envía el cerebro al cuerpo. El mensaje de movimiento no puede llevarse a cabo mediante el uso del músculo esquelético para crear el movimiento voluntario. Como los músculos del cuerpo no reciben mensajes de los nervios, se atrofian y se encogen con el tiempo. El movimiento de la cara, lengua, garganta, brazos, pecho y piernas se ve especialmente afectado. Esto afecta las funciones de respirar, tragar, hablar y caminar.
Las funciones corporales que no se ven afectadas por la SMA incluyen funciones como el aprendizaje y las habilidades sociales. Los nervios sensoriales no se afectan y por lo tanto, se mantienen las sensaciones o impresiones en el cuerpo. La vista y la audición no se ven afectadas. Las funciones de los intestinos y de la vejiga tampoco se ven afectadas.
Genética
La SMA comúnmente es provocada por una mutación o por la falta de un gen, lo que la convierte en una enfermedad genética (heredada). La SMN es una proteína del cuerpo necesaria para la supervivencia y la deficiencia de esta tiene como resultado la pérdida de la neurona motriz selectiva. El gen afectado que provoca la SMA se conoce como el gen 1 (SMN1), gen de supervivencia de las neuronas motrices, que se localiza en el cromosoma 5q. Este gen es responsable de mantener la salud y la función de las neuronas motrices, que son los nervios que generan el movimiento en el cuerpo. El gen SMN2 le da la instrucción al cuerpo de que forme el gen SMN1. Si SMN1 está ausente y SMN2 falta, la SMN no se puede producir. Las personas con falta de SMN1 tienen SMN, aun cuando algunas personas con falta de SMN1 pueden confiar en SMN2 para la producción de la SMN.
Para tener la SMA, las personas necesitan heredar un gen recesivo (no dominante) de ambos padres. Heredar solo un gen recesivo de uno de los padres no produce la enfermedad, pero la persona será portadora de SMA. Ha habido casos raros en los que los factores genéticos no están claros.
Hay muchas dudas acerca de la expectativa de vida en las personas con SMA. Tradicionalmente, el tipo más bajo de SMA tiene una menor expectativa de vida. El tipo mayor IV tiene una expectativa de vida que es la misma a la de la población en general. Sin embargo, con el desarrollo de los nuevos tratamientos para mejorar la SMA, las expectativas de vida se están incrementando. Asimismo, los investigadores están aumentando constantemente las opciones para mejorar la respiración, la movilidad y otras complicaciones de la SMA.
Tipos de SMA
Existen cinco tipos de SMA que se basan en la mejor función motriz. La Asociación de Distrofia Muscular (MDA, por sus siglas en inglés) estableció un sistema de clasificación de la SMA en 1991. El grado de gravedad varia dentro de cada tipo. Los tipos se dan a través de las categorías. Solo alrededor del 25% de las personas con SMA se pueden clasificar claramente dentro de un solo tipo. Podrá oír que se está utilizando este sistema de clasificación de “tipo”.
Sistema original de clasificación de SMA
| Tipo de SMA | Edad de diagnóstico/aparición | Signos clínicos | Capacidad |
| SMA tipo 0 | Prenatal | · Movimiento fetal reducido · Malos reflejos o falta de ellos · Parálisis facial · Defecto del tabique interventricular · Contracturas articulares y problemas esqueléticos · Espasmos musculares · Dificultad para tragar y para alimentarse · Insuficiencia respiratoria | Se requiere apoyo de respiración |
| SMA tipo I También conocida como enfermedad Werdnig-Hoffmann | 0-6 meses | · Bajo tono corporal (postura de pata de rana) · Mal control de la cabeza · Malos reflejos o sin respuesta · Respiración desde el estómago por poca fuerza en los músculos intercostales (entre las costillas), pero el diafragma funciona bien · Riesgo de aspiración debido a debilidad de la motricidad oral · Actividad mental sin afectar | No puede rodar ni sentarse por sí mismo |
| SMA tipo II | Menos de 18 meses | · Normalmente, debilidad progresiva en las piernas pero no tanto en los brazos · Bajo tono, sin reflejos · Complicaciones óseas debido a músculos débiles como contracturas por escoliosis, rigidez en la mandíbula · Enfermedad pulmonar restrictiva · Actividad mental sin afectar | Capacidad para sentarse pero no para estar de pie/caminar |
| SMA tipo III También conocida como enfermedad de Kugelberg-Welander | Mayor de 18 meses, Subtipos: 3a 18 meses – 3 años 3b >3 años | · Debilidad en las piernas mayor que en los brazos · Poca debilidad muscular al respirar o escoliosis · Dificultad para correr, subir escaleras o intentar ponerse de pie · Actividad mental sin afectar · Expectativa de vida sin afectar | Camina pero puede demorarse |
| SMA tipo IV | Mayor de 21 años o en la edad adulta | · 5% de los casos · Aparición en la edad adulta, muy a menudo alrededor de los 30 pero puede ser antes o después · Debilidad leve a moderada en las piernas · Actividad mental sin afectar · Expectativa de vida sin afectar | Camina de forma independiente o con dispositivos de movilidad asistida |
| SMA no relacionada con el cromosoma 5 (SMN1 o proteína de SMN) | Varía | · Varía en gravedad · Puede incluir músculos lejos del centro del cuerpo | Varía |
Sistema actual de clasificación de la SMA
La SMA es un problema de salud que cambia con el tiempo. A algunas personas se les clasifica conforme a lo anterior mientras que otras van de un tipo a otro con base en su capacidad funcional. El cambio puede implicar mejoras o pérdidas de función dependiendo de cada persona. Por lo tanto, se desarrolló un sistema de clasificación más práctico para indicar el nivel actual de la función en lugar de la función en el momento del diagnóstico. Este sistema de clasificación se divide en tres categorías y se agregan subcategorías a medida que se investiga más. Las clasificaciones son: los que no se sientan, los que se sientan y los que caminan. Nadie se ajusta perfectamente a una sola categoría y los cambios de categoría son frecuentes.
| Categoría | Clasificaciones de tipo | Características |
| Los que no se sientan | En su mayoría tipo I | · Dificultad para respirar y para alimentarse · Deformidad del tórax con forma de campana · Fasciculación de la lengua · Flexión de cadera ausente, dejan caer la cabeza |
| Los que se sientan | En su mayoría tipo II y III | · Logran sentarse pero no caminar · La debilidad muscular es más profunda en las piernas que en los brazos · Se desarrollan contracturas articulares, anquilosis de mandíbula y escoliosis con frecuencia · la mayoría no tienen problema para respirar ni para tragar; sin embargo, la debilidad de los músculos intercostales puede provocar enfermedad pulmonar restrictiva e insuficiencia respiratoria La SMA puede avanzar entre las edades de 5 y 15 años, especialmente durante la pubertad |
| Los que caminan | Tipo II o IV | · Los que logran o conservan la capacidad para caminarMenor función de las piernas que debilidad en los brazos Normalmente, no tienen dificultad para respirar ni para tragar; sin embargo, a lo largo de la vida, el caminar y respirar pueden deteriorarse lenta y levemente · El 95% de los que se sientan pronto pueden caminar a los 18 meses · El 50% de los que se sientan tarde pueden caminar a los 18 meses · La expectativa de vida es igual a la de la población en general |
| Aparición en Adultos | Tipo IV | · Pueden aparecer síntomas como incomodidad o dolor en los músculos de las piernas · Las piernas pueden debilitarse y requerir la colocación de férulas o aparatos ortopédicos para poder seguir caminando · Normalmente tragar y respirar no se ven afectados |
Síntomas/Diagnóstico
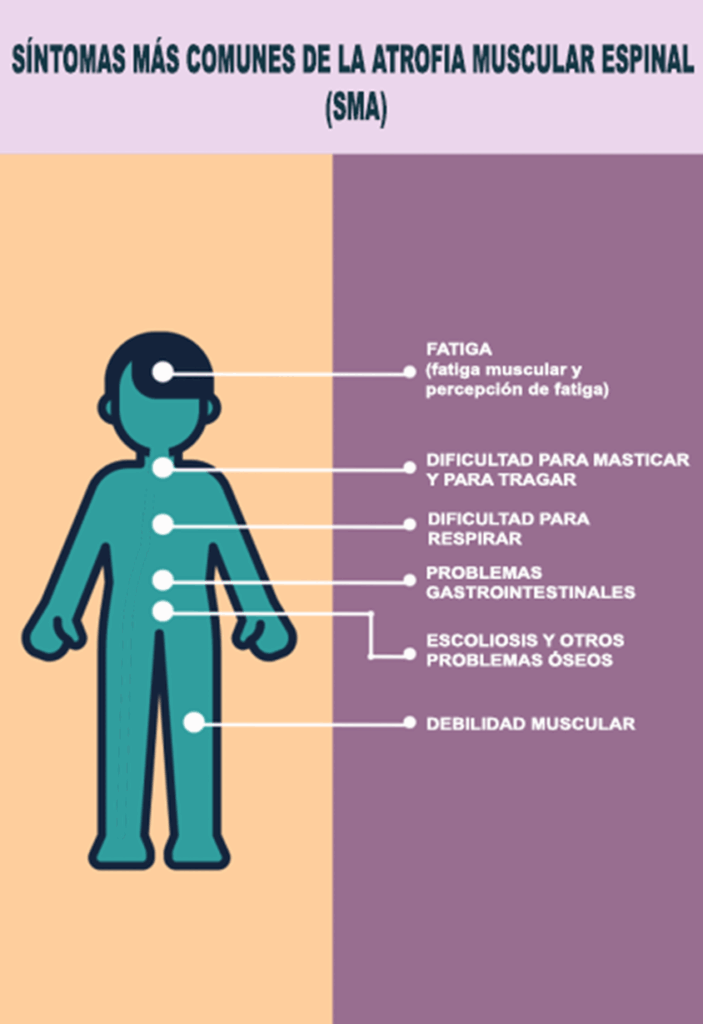
Generalmente la debilidad muscular es el primer síntoma de la SMA. Si la debilidad muscular es severa, especialmente con más debilidad en el centro del cuerpo y menos en las extremidades, el diagnóstico estará bastante claro. La debilidad leve puede no relacionarse directamente con el diagnóstico de SMA.
Las pruebas genéticas que utilizan una muestra de sangre para evaluar si existen mutaciones o supresiones en los genes SMN1 y SMN2, representan la prueba definitiva. También se podrá detectar a una persona portadora de SMA. Si un varón y una mujer son ambos portadores, pueden optar por tomar una decisión personal y utilizar a un donador para reducir el riesgo de que el bebé adquiera la enfermedad. La prueba genética de SMA tiene una confiabilidad del 95%.
La electromiografía (EMG) o prueba de la función muscular registra la actividad eléctrica de los músculos grabando la actividad eléctrica de estos y se puede hacer comparando la contracción y la relajación de los músculos.
Los estudios de conducción nerviosa (NCS, por sus siglas en inglés) pueden realizarse en el momento de la EMG. Esta prueba evalúa la capacidad de un nervio de transmitir mensajes.
Este gráfico se reimprimió con el permiso de SMA News Today.
La biopsia muscular puede evaluarse cuando se retira un muy pequeño fragmento de un músculo del cuerpo y se examina microscópicamente para ver cambios que indiquen enfermedad neuromuscular.
Biomarcadores: Los cambios en ciertas características del cuerpo o de la sangre, conocidos como biomarcadores, pueden indicar enfermedades. El número de copias de SMN2 se correlaciona inversamente con el diagnóstico de SMA y con la indicación de gravedad. Las medidas de EMG de los potenciales compuestos de acción muscular, el cálculo de unidades motrices y la miografía de impedancia eléctrica, predicen la SMA y calculan los medicamentos para su tratamiento.
Otras pruebas de sangre incluyendo SMN mRNA y los niveles de proteína, pueden revelar los materiales de desecho resultado de la destrucción muscular.
Tratamiento
Hay medicamentos disponibles para diferentes tipos específicos de SMA.
Nusinersen (SPINRAZA) aumenta la producción de la proteína SMN mediante la modulación del corte y empalme de SMN2 lo que mantiene a las neuronas motrices. Este medicamento se administra intratecalmente (en el espacio entre la médula espinal y la membrana que la contiene) mediante una inyección en la columna vertebral. Está disponible para niños y para adultos con cualquier tipo de SMA.
Onasemnogene abeparvovec-xioi (Zolgensma) es una terapia de reemplazo del gen SMN1. La producción de la proteína SMN se mejora mediante la transferencia del gen SMN1 que entra a las células neurales (de los nervios) utilizando estereotipos del virus adeno-asociado. Estos virus no son patógenos (no enfermarán a su hijo(a). Pasan fácilmente a las células que se utilizan comúnmente en la terapia de genes por su seguridad. Hay más información en el Pillay, artículo de 2017 en la sección de referencia. Está disponible con relación a niños menores de dos años con aparición infantil de SMA. El objetivo consiste en mejorar el movimiento y la función muscular así como la supervivencia.
Risdiplam (Evrysdi) es un medicamento oral para estimular la expresión del gen SMN3 de la proteína SMN. Es para niños de dos años y mayores.
La cantidad de estudios de tratamientos de SMA ha sido abundante desde el descubrimiento del gen SMN. Esto ha conducido a estudios de laboratorio así como a estudios clínicos (con humanos) de forma constante. Estos estudios incluyen terapia con pequeñas moléculas para aumentar la proteína SMN, la terapia basada en RNA para aumentar el SMN2 en la molécula de ARN para un aumento en la expresión de SMN2, y la terapia de genes para sustituir el gen SMN1 faltante.
Se están llevando a cabo más estudios de forma rápida. Consulte el sitio web: https://clinicaltrials.gov/ para ver nuevos ensayos clínicos patrocinados por el gobierno de los EE. UU. La información de ensayos clínicos individuales de SMA le brindará conocimiento de las ideas de los investigadores así como de los ensayos más recientes. Es importante saber lo que sucederá en el futuro para poder prepararse y tomar decisiones bien informadas en torno a la salud de su hijo(a).
Terapias de Rehabilitación
Un equipo de profesionales de la salud se encargará del cuidado de su hijo(a). Cada uno aporta su especialidad para satisfacer las necesidades individuales trabajando al unísono.
El diagnóstico está a cargo de un genetista o de un neurólogo a quien se consulta en caso de que haya sospecha de SMA. Puede resultar una experiencia agotadora. Es útil contar con información de la enfermedad para entender las opciones de tratamiento médico que están disponibles para su hijo(a) y para mantener su calidad de vida y sus experiencias. Esta persona contribuirá al Programa o Plan de Educación Individualizado (IEP, por sus siglas en inglés), que será necesario llevar en la escuela para satisfacer las necesidades específicas del menor. También podrán ayudar con la planificación familiar. Le darán opciones de terapias que podrá investigar en caso de que le interese seguir esta vía de tratamiento.
Un psicólogo puede mejorar el bienestar mental trabajando con los menores y con su familia. Esta persona es útil para explorar las emociones y los efectos que esto tiene en su hijo(a), en su familia y en usted. También contribuirá al Programa de Educación Individualizada (IEP) que deberá estar presente en la escuela para satisfacer las necesidades específicas del menor.
Los especialistas en trastornos pediátricos neuromusculares, un fisiatra pediátrico o un fisiatra para adultos (dependiendo de la edad) le proporcionarán atención médica especializada. El médico se encargará de las necesidades funcionales de su hijo(a) para promover la enseñanza de habilidades (aprender nuevas habilidades) y la rehabilitación (volver a aprender las habilidades). Esta persona puede ser el líder del equipo de atención médica quien organizará y proporcionará atención que incluirá tratamiento médico, equipo, terapia, medicamentos y pruebas. Estarán conscientes de la investigación y conocerán los nuevos tratamientos que le aportarán beneficios.
Puede ser necesario que los educadores de escolarización como el director de la escuela, los maestros y los auxiliares educativos apoyen el aprendizaje de su hijo(a). Involucrar al personal de la escuela mediante el uso del IEP es fundamental. Con la SMA, la capacidad de aprendizaje no se ve afectada, pero los requisitos motrices para respirar y el equipo de movilidad, tomar notas, la actividad física, ayuda con alimentación, tiempo de descanso y otros problemas pudieran requerir un equipo adaptado. El IEP es un plan creado y documentado con relación a las necesidades de su hijo(a) y a la forma como el sistema escolar cumplirá con ellas.
Los terapeutas físicos, ocupacionales y logopedas/foniatras recomendarán equipo para lograr la máxima independencia posible. Es necesario contar con equipo de posicionamiento para mantener el cuerpo alineado, lo que apoya la respiración, el sistema esquelético y la digestión, entre otras funciones. El equipo puede incluir sistemas de descanso, para sentarse, sillas de ruedas, caminadoras, aparatos ortopédicos y equipamiento de baño.
Las terapias para la atrofia del músculo espinal se basan en las necesidades individuales. No todas las personas necesitan todos los tratamientos; sin embargo, saber que están disponibles es útil para mejorar la salud y para evitar problemas que empeoren la salud.
Respiración: Un neumólogo pediátrico o de adultos y un terapeuta respiratorio se encargarán de recetar y de coordinar los tratamientos respiratorios necesarios. Estos pueden incluir ventilación mecánica que pudiera requerir una traqueostomía, ventilación mecánica no invasiva que no requiere traqueostomía, succión, insuflación, tratamientos con nebulización y ejercicios de respiración como espirometría estimulada. Las infecciones respiratorias pueden ocurrir con mayor facilidad, por lo tanto, es importante lavarse las manos, hacer ejercicios de respiración, llevar a cabo el distanciamiento social y el aislamiento de personas enfermas.
Algunos menores pueden padecer de apnea profunda durante el sueño. Deberá llevarse a cabo una evaluación y monitorear los síntomas como el ronquido, el resoplido, periodos sin respirar o el dolor de cabeza por la mañana al estar acostado. Los problemas de respiración pueden provocar otros padecimientos ya que pueden afectar la función cardiaca.
Las motricidades orales pueden fortalecerse y mejorarse mediante la ayuda de un logopeda/foniatra que le ayude a masticar, tragar y a articular mejor las palabras. En el caso de que tragar resulte un problema, pueden utilizarse métodos alternos de alimentación como alimentación por sonda.
Los problemas de motricidad oral en la comunicación pueden interrumpir esta última. Hay dispositivos disponibles que ayudan a comunicarse y requieren un movimiento ligero de las manos o el uso de tecnología de fijar la mirada.
Nutrición: Los problemas de masticar, tragar y llevarse alimentos a la boca físicamente pueden ser difíciles. Es necesario garantizar la nutrición adecuada. Puede resultar difícil mantener la ingesta calórica para la salud ósea, el crecimiento y el desarrollo o evitar la sobrealimentación. La altura, el peso corporal y el índice de masa corporal (IMC) deben monitorearse de forma continua. Un especialista en nutrición puede ayudar a proporcionar una dieta adecuada que funcione bien para su hijo(a). Un terapeuta ocupacional puede ayudar con el uso de equipo adaptado.
Problemas gastrointestinales: Los problemas para masticar, las dietas líquidas y el movimiento reducido pueden hacer que el sistema digestivo funcione con más lentitud. Esto puede provocar reflujo gástrico, sentirse lleno(a) por el vaciado lento del estómago o por la función intestinal, vómito y estreñimiento. Los problemas metabólicos, la presión alta, el azúcar en sangre y la descomposición de grasas pueden ser un desafío. El ejercicio mejora la digestión y el metabolismo de la glucemia. La terapia de estar de pie también ayuda a la digestión. Los gastroenterólogos se especializan en problemas gastrointestinales. Puede ser necesario tomar medicamentos para los problemas anteriores.
Ortopedia: Los problemas del esqueleto son un desafío cuando se padece SMA. Un ortopedista, el profesional médico que se encarga de los huesos, ayuda no solo a tratar los problemas óseos sino también a evitar dichos problemas.
Estirar las articulaciones del cuerpo es importante para mantenerlas flexibles. Es una actividad que puede hacer tu hijo(a) con ayuda en caso de que no pueda mover el cuerpo de manera independiente. Esto ayuda a evitar contracturas y calambres y proporciona alivio general al cuerpo.
Pueden utilizarse férulas y aparatos ortopédicos para mantener en cuerpo en la posición correcta, para estabilizar las articulaciones y para promover el movimiento. Pueden utilizarse en brazos, piernas, torso, cuello o donde se necesite corregir la postura. Algunos se llevan solo por las noches.
Las contracturas o la rigidez de las articulaciones es un problema común con la SMA ya que el movimiento representa un desafío. Se proporcionará y se enseñará el tratamiento terapéutico al menor y a los miembros de su familia para que pueda mantener o mejorar la flexibilidad y hacer más lento el avance del desgaste muscular y para que pueda moverse y hacer ejercicio. Se podrán reducir las contracturas con cirugía ortopédica o con yesos seriados liberando la articulación con el tiempo.
La posición es importante para el mantenimiento y la función del cuerpo. El equipo para alinear el cuerpo anatómicamente ayudará a mantener el cuerpo en forma eficaz y funcional. Los ejercicios de estiramiento, las férulas y los yesos seriados también pueden ayudar.
Los bipedestadores ayudan a asumir una posición recta que permite que el menor vea el mundo desde una altura natural, mejoran las interacciones personales en algunas situaciones, mejoran el desarrollo óseo y pueden ayudar con la postura y la digestión. Consulte lo anterior con el profesional de la salud de su hijo(a) si el menor está listo físicamente para esta actividad, para evitar complicaciones inesperadas, especialmente en los huesos o en el sistema vascular.
La escoliosis (columna vertebral en forma de ‘S’) y la cifosis (inclinación de la columna hacia el frente en forma de ‘C’) son problemas por la debilidad en los músculos del tronco. Los desequilibrios musculares pueden jalar las vértebras de la columna y desalinearlas, reduciendo la capacidad para respirar de manera eficaz. La escoliosis puede manejarse con ejercicio, aparatos ortopédicos e intervención quirúrgica en caso necesario. En el caso de que se requiera la intervención quirúrgica, puede tener la opción de estabilización rígida de la columna o de estabilización flexible. Las nuevas varillas de estabilización flexible permiten cierto movimiento ligero del tronco una vez que ha sanado.
La alienación de la cadera es un elemento crítico cuando se está en cama y cuando se está sentado(a). Hay que comprobar que las caderas estén alineadas y que haya estiramiento de los músculos de la cadera y eso mantendrá el cuerpo en buena postura que evitará problemas de contracturas, mala alineación de cadera y escoliosis.
La movilidad y el ejercicio son críticos para todas las personas. El cuerpo ansía el movimiento. El ejercicio beneficia la respiración, la digestión y la salud mental y general. Los dispositivos de movilidad como las sillas de ruedas, las caminadoras, las férulas y los aparatos ortopédicos le ayudarán a su hijo(a) a moverse en su entorno. Esto crea independencia en su mundo cuando están listos para ello. El ejercicio mediante la terapia se receta a nivel individual. Otras opciones de ejercicio pueden incluir juegos de movimiento, terapia acuática, deportes adaptados o cualquier cosa que ayude a que el cuerpo funcione.
Salud corporal: Cuando los huesos no sostienen el peso corporal, los minerales tienden a reducirse haciendo que los huesos se vuelvan frágiles y eso tiene como resultado las facturas de hueso. La osteopenia es la pérdida de densidad ósea y esto lleva a la osteoporosis. Deben realizarse pruebas de densidad ósea anualmente en las personas con SMA de todas las edades para conocer la densidad ósea de forma temprana para poder iniciar su tratamiento. El ejercicio y estar de pie en un bipedestador ayudan a mantener la densidad ósea pero hay algunos casos más severos que requieren medicamentos.
El equipo de cuidados paliativos consta de un grupo de personas, incluyendo a enfermeros especializados, que brindan ayuda médica para las necesidades de calidad de vida. Este servicio no es solo para cuidados al final de la vida sino que garantizará que se satisfagan las necesidades de su hijo(a) con apoyo. La SMA puede evolucionar y adoptar diferentes vertientes. Este grupo puede estar al cuidado de su hijo(a) o dejar de estarlo, conforme cambien las necesidades.
Historia
1891: La SMA se describió por primera vez en la literatura médica cuando Gundo Werdnig descubrió el padecimiento en dos hermanos pequeños. Posteriormente, Johan Hoffmann describe la debilidad progresiva en menores de 1893 a 1900, que fue cuando se utilizó por primera vez el término “atrofia del músculo espinal”. Werdnig-Hoffmann se convirtió en el nombre que se utilizaba para la SMA tipo 1; sin embargo, sus descripciones eran de casos intermedios.
Se presentaron casos severos en 1899 y en 1903. Los casos más leves de personas con SMA capaces de caminar y de estar de pie, se presentaron en la década de los años cincuenta. Había cierta controversia en los tipos y en las clasificaciones y fue hasta 1991 cuando la Asociación de Distrofia Muscular patrocinó el sistema de clasificación en el Consorcio Internacional de Atrofia Muscular de la Columna Vertebral con base en los tres tipos de SMA y en la edad de aparición.
No fue sino hasta 1995 cuando el laboratorio Melki determinó que la causa de la SMA era la supresión del gen SMN1 en el cromosoma 5Q13 y SMN2. Ese fue un descubrimiento fundamental ya que antes de este, no se conocía ninguna razón específica para el desarrollo de la SMA; por lo tanto, establecer un tratamiento era impensable. Cinco años después del descubrimiento genético, se pudieron empezar a crear modelos animales para estudiar la SMA y empezar a desarrollar el tratamiento para esta enfermedad.
En 2007, Wang y sus colegas desarrollaron la primera pauta clínica para el cuidado de personas con SMA, incluyendo el envejecimiento con la enfermedad.
Desde que se entendieron los problemas genéticos, el Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrales, varias fundaciones, farmacólogos y biotecnólogos empezaron a desarrollar terapias para aumentar o mejorar el SMN1 y el SMN2 utilizando terapia de pequeñas moléculas, terapia ARN y terapia de genes. Ha habido resultados significativos con los siguientes tratamientos:
- 2016 – La FDA aprobó el tratamiento Nusinersen (SPINRAZA) para todos los tipos de SMA.
- 2017- La FDA aprobó el tratamiento Onasemnogene abeparvovec-xioi (Zolgensma) con enfoque en el reemplazo del gen SMN1.
- 2020 – La FDA aprobó el tratamiento Risdiplam (Evrysdi) para estimular la expresión del gen SMN2 de la proteína SMN. Este fue el primer medicamento oral para el tratamiento de SMA.
La investigación ha aumentado constantemente y se han llevado a cabo más estudios en diferentes condiciones de los ensayos clínicos. Asimismo, es importante recordar que muchos otros investigadores han ayudado a mejorar la vida de las personas con SMA gracias a su investigación de las mejoras en la respiración, en los medicamentos y en el tratamiento de escoliosis y de otros padecimientos óseos, así como al enfoque en los problemas de calidad de vida, entre muchos otros.
Datos y Cifras
SMA es la causa más común de muerte debido a la alteración genética en los niños en la primera infancia.
Uno de cada 6,000 bebés nace con SMA. Una de cada 11,000 personas de todas las edades se ven afectadas por la SMA.
Los portadores de la anomalía genética de la SMA que no padecen la enfermedad son 1 de cada 54.
El tipo más común de SMA es la enfermedad de Werdnig-Hoffmann, que es responsable del 80% de los casos. La enfermedad de Kugelberg-Welander es otro tipo de padecimiento que se diagnostica en la primera infancia.
El desarrollo de SMA en la primera infancia y en la niñez se traduce en peores resultados que si se desarrolla en una etapa posterior.
Recursos
Si desea obtener más información de la atrofia muscular de la columna vertebral o si tiene alguna pregunta específica, nuestros especialistas en información están disponibles de lunes a viernes. Puede llamar sin costo al 800-539-7309.
