Leukodystrophy
The brain and spinal cord consist of substances called white matter and gray matter. Gray matter is responsible for information processing, muscle movement, sensory perception, memory, emotions, and overall brain functions. It consists of neuronal cell bodies, dendrites, and axons (all parts of nerves). White matter is responsible for nerve communication and signal transmission from gray matter to areas of the brain and spinal cord. White matter consists mostly of axons (the long arm of a nerve cell that transmits messages from one cell to another) coated or insulated in a fatty sheath called myelin.
Differences between the brain gray and white matter
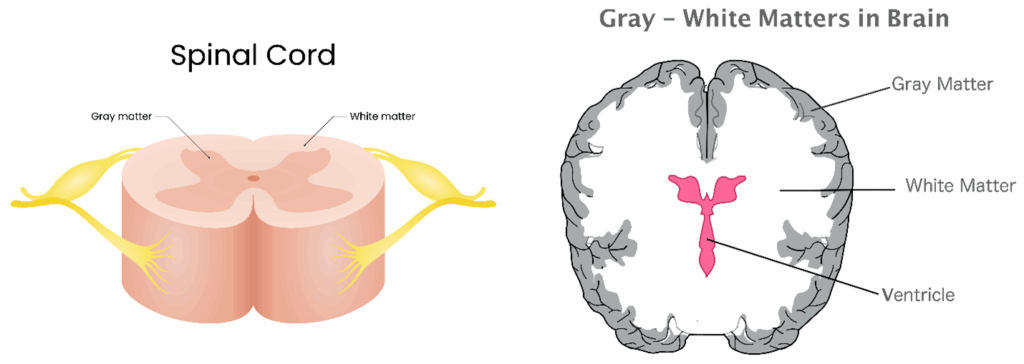
The unmyelinated nerves of gray matter have a gray coloring; myelin has a whiteish color, which is why these two anatomical divisions are called gray and white matter.
In the brain, gray matter is in the outer layers. In the spinal cord, gray matter is shaped like a ‘butterfly’ in the center of the spinal cord. In the brain, gray matter surrounds white matter, but in the spinal cord, white matter surrounds gray matter.
Actions of Gray and White Matter Work Together
| Brain | Spinal Cord | |
| Outer layers | Gray matter is in the outer layers | White matter is in the outer part of the cord |
| Inner layers | White matter is in the inner part of the brain | Gray matter is a ‘butterfly’ shape in the center of the cord |
| Comprises | Gray matter is mostly neuronal cell bodies | White matter is mostly sensory and motor pathways |
| Activity performed | Gray matter processes information | White matter transmits signals to and from the brain |
Damage can occur to the myelin sheath (covering) of white matter nerves in the brain and spinal cord due to genetic issues. Genetic changes occur from inherited genes or spontaneously. Leukodystrophy can arise from a gene mutation, a slowing, or an absence that affects functioning to maintain health. Most leukodystrophies are monogenetic, meaning only one gene is affected. Other forms of leukodystrophy involve multiple genes. As researchers learn more about the human genetic code, more information about leukodystrophy will be discovered.
Leukodystrophy is a collection of genetic diseases affecting white matter in the brain and/or spinal cord. The specific gene or genes affected determine the type of leukodystrophy. These gene mutations predict the onset, severity, and progression of the disease. This damage can slow or block the transmission of nerve signals, affecting functions such as movement, vision, hearing, and cognitive function, among other bodily functions. Some forms of leukodystrophies overlap in genetic causes and symptoms, which makes diagnosis more challenging.
There are three basic types of leukodystrophies:
• Demyelinating leukodystrophies damage myelin, the insulation around nerve fibers in white matter.
• Hypomyelination leukodystrophies do not produce enough myelin to surround the nerves.
• Leukoaxonopathies lead to damage to myelin and the nerve.
Currently, there is no cure for leukodystrophy. Scientists and researchers are gaining a deeper understanding of human gene function, as well as the disease itself. Progress is being made in medical treatments. Some therapies are available to assist with adaptation and perhaps delay progression.
What are the Types of Leukodystrophy
Over 58 types of leukodystrophies have been identified. Each of these types develops due to mutations in different genes, resulting in distinct symptoms. This list includes the more common types. Types may also have subtypes.
| Type/Gene Mutation | Onset | Health Issues |
| Adrenoleukodystrophy (ALD) Affects the myelin of the brain, spinal cord, and affects the function of the adrenal glands (hormone production) Gene mutation is on the ABCD1 X chromosome. This results in neurological issues and loss of function. | Childhood usually around age 7 Sometimes in early adulthood | Learning issues Vision and/or hearing loss Dysarthria (unclear speech) Imbalance Lack of coordination Fatigue Weight loss Cognitive and behavioral issues |
| Adult-onset autosomal-dominant leukodystrophy (ADLD) Inherited from a dominant gene resulting in white matter damage. ADLD may have more than one causative mutation. The involved genes will produce different neurological effects. | 40-50 years | Autonomic dysfunction (automatic) functions, such as heart rate and blood pressure, among others Strength Movement Cognition |
| Adult Polyglucosan Body Disease A mutation in the GBE1 gene that codes for the glycogen branching enzyme. Neurological effects are prevalent. | 30-40 years | Weaknesses and sensory loss especially legs and arms Walking challenges Gait instability Bladder and bowel issues Cognitive issues, memory loss, and confusion Dementia |
| Aicardi-Goutières Syndrome (AGS) Occurs from mutations in several genes (from both parents), leading to damage in the brain white matter. Affected are the development and function of the brain and immune systems. | Early Childhood | Microcephaly (small head) Seizures Developmental delays Intellectual challenges Motor coordination issues Eye abnormalities Skin rashes Liver function issues Immune system dysfunction |
| Alexander disease Mutation occurs in the Glial Fibrillary Acidic Protein (GFAP) gene, creating Rosenthal fibers, leading to astrocyte degeneration and demyelination. Leading to the disruption of nerve function and communication. | Infant onset: Birth-2 years Childhood onset: Between 4-10 years Adult onset: Later adult years | Infant onset: Developmental delays Macrocephaly (large head) Seizures Walking challenges Swallowing issues Childhood onset: Motor function decline Cognitive issues Seizures Adult onset: Same as childhood onset Psychiatric issues |
| CADASIL (Cerebral Autosomal Dominant Arteriopathy Infarcts and Leukoencephalopathy) It affects the brain blood vessels caused by a mutation in the NOTCH3 gene. | Adulthood | Small strokes Cognitive decline Dementia Migraines with aura Mood changes Seizures |
| Canavan disease A mutation deficiency of the ASPA gene on chromosome 17p13.2. This leads to an accumulation of N-acetyl-L-aspartic acid (NAA), causing oligodendrocyte (a type of nerve cell) dysfunction and myelin degradation. | Primarily in infancy Some later in life Prevalent in Ashkenazi Jews, but also occurs in other populations | Developmental delay Weaknesses Swallowing difficulty Seizures Irritability Vision changes |
| CARASIL (Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) Damage to small blood vessels leading to stroke-like episodes caused by a mutation in the HTRA1 gene. | 20-50 years | Early-onset stroke-like episodes Spasticity Pseudobulbar palsy, including difficulty chewing, swallowing, and speaking Spine changes Dementia Gait disturbances Premature hair loss |
| CerebroTendinous Xanthomatosis (CTX) Mutations in the CYP27A1 gene, which is responsible for the breakdown of cholesterol. It affects white matter leading to issues in the brain, tendons (attach muscles to bones), heart, and eyes. | Childhood with increased issues in adulthood | Thinking issues, including dementia, hallucinations, seizures, and ataxia Walking difficulty from tendon issues Eye problems, including cataracts Heart disease Osteoporosis Liver issues |
| Childhood ataxia with central nervous system hypomyelination (CACH) (Vanishing white matter disease) Mutation of genes for the eIF2B translation initiation factor leading to an inability to regulate protein synthesis. Destroys white matter in the brain and spinal cord. | Early childhood | Developmental delay Optic atrophy (vision loss) Speech loss Seizures Walking challenges Balance issues Spasticity |
| Fabry disease A mutation in the GLA gene affects the body’s ability to break down glycosphingolipid fat, causing it to collect in the cells. | Onset in childhood or adolescence Late onset in adulthood (30+) | Pain in hands, feet, and limbs Skin rashes Diarrhea, bloating Heat intolerance Sweating decreased Later developments: kidney, heart, stroke, and neurological issues |
| Fucosidosis A deficiency in the alpha-L-fucosidase enzyme causes a buildup of fucose-containing sugars in cells. | Type I rapid progression in childhood, early death Type II slower progression with survival into adulthood | Intellectual disability Motor skill delay Seizures Respiratory infections Characterizations: Prominent forehead, broad and flattened nose, thickened lips and tongue, large, low-set ears. |
| GM1 Gangliosidosis A mutation of the GLB1 gene leads to a lysosomal storage disorder with the accumulation of a fatty substance (GM1 ganglioside) in the brain and other tissues. | Infantile GM1 gangliosidosis: the most severe form, presenting in the first few months of life. Juvenile GM1 gangliosidosis: presents in childhood with milder symptoms Late-onset GM1 gangliosidosis: the rarest form, presenting in adulthood | Infantile GM1 gangliosidosis: Developmental regression, Seizures Enlarged liver and spleen Cherry-red spots in the eyes. Juvenile GM1 gangliosidosis: Cognitive decline Unsteady gait Muscle weakness. Late-onset GM1 gangliosidosis: symptoms similar to juvenile GM1 gangliosidosis, but less severe. |
| Krabbe disease (globoid cell leukodystrophy) A genetic mutation in the GALC gene leads to a buildup of toxins in myelin. | Infancy or beyond | Weaknesses Feeding challenges Irritability Sensory deficits Developmental delay Neuropathy Spasticity Hypotonia Myoclonic Seizures |
| L-2-hydroxyglutaric aciduria Mutations in the L2HGDH gene, located on chromosome 14q22.1, leading to hydroxyglutarate compound accumulation in the body with toxic effects on the central nervous system (CNS). | Most often between 1-3 years but can be earlier or later | Developmental delay Seizures Muscle weakness Cerebrum abnormalities affecting speech, vision, emotion, memory, and thinking Macrocephaly (large head) |
| Megalencephalic Leukoencephalopathy with Subcortical Cysts Genetic mutations in either the MLC1 gene (in about 75% of cases) or the HEPACAM gene (in about 20% of cases). | Typically, the first year of life | Macrocephaly (enlarged head) Motor development delay Spasticity Seizures |
| Metachromatic leukodystrophy Genetic mutation of the arylsulfatase A (ARSA) gene on chromosome 22q13.3-qter, resulting in an accumulation of sulphatide (a multifunctional molecule) affecting the nervous system. | Two years or younger Some in childhood and as an adult | Ataxia Developmental delay Thinking issues Behavior changes Vision loss or blindness Hearing changes Decerebrate posturing Motor skill challenges Seizures Neuropathy Dementia |
| Multiple Sulfatase Deficiency Mutations in the SUMF1 gene make the body unable to break down sulfates (complex sugars). | Prebirth form is the most severe Late infancy (birth to 2 years) is the most common | Developmental delays Intellectual challenges Seizures Hearing loss Vision problems Joint stiffness Bone issues Coarse facial features Respiratory issues Liver and kidney dysfunction |
| Pelizaeus-Merzbacher disease (PMD) A mutation of the proteolysis protein 1 (PLP1) gene on the X chromosome leads to neurological issues and developmental delays. | The onset detected in the newborn is more severe. Has an adult form with less severity. Affects mostly males. | Nystagmus (involuntary eye movements) Ataxia Coordination issues Cognitive issues Weaknesses Motor skills issues Spastic paralysis Developmental delays Seizures Respiratory issues |
| Pol III-Related Leukodystrophies A spectrum of neurodegenerative disorders caused by genes that encode subunits of the RNA polymerase II enzyme. POLR3A: Encodes the largest catalytic subunit of the Pol III enzyme. POLR3B: Encodes the second-largest catalytic subunit. POLR1C and POLR3K: Encodes smaller, accessory subunits of the Pol III complex. | Typically, early childhood (1-5 years), and less frequently, anytime in the lifespan | Motor issues (balance and walking) Speech challenges Body posturing Spasticity Learning challenges Abnormal eye movement Chewing and swallowing issues Less teeth Short stature Hormonal delay Near sightedness Cataracts Optic atrophy |
| Refsum disease A mutation of the PHYH or PEX7 genes leading to a deficiency in the enzyme phytanoyl-CoA hydroxylase, causing an accumulation of phytanic acid (a fatty acid) in the body. | Appears at any age, mostly in adolescents and adults. | Vision issues and loss Imbalance Walking challenges Numbness/Tingling in hands and feet Skin dryness Heart rhythm abnormalities Liver issues Kidney issues Bone issues |
| Salla Disease (free sialic acid storage disease) A genetic mutation in the SLC17A5 gene is characterized by a progressive buildup of sialic acid inside cells. This affects cell signaling, immune response, and brain function. | Typically, 6-12 months, mildest form Less often, 1-6 months, intermediate form Prebirth is the most severe form | Intellectual disability Delayed development Poor muscle tone Symptoms worsen over time |
| Sjogren-Larsson Syndrome Mutations in the ALDH3A2 gene, which encodes an enzyme, fatty aldehyde dehydrogenase (FALDH). FALDH is involved in the breakdown of fatty aldehydes, leading to an accumulation in the body. | Dry skin is apparent at birth, with diagnosis made by 3 years | Dry, scaly skin Spasticity Seizures Intellectual disability Dry eyes Cataracts Optic nerve damage Short stature Joint stiffness Motor development delays |
| X-linked adrenoleukodystrophy A mutation in the ABCD1 gene, which provides instructions for creating a protein, adrenoleukodystrophy protein (ALDP), a transporter that helps move very long-chain fatty acids (VLCFAs) into cellular sacs called peroxisomes for breakdown. The result is progressive neurological issues and adrenal insufficiency. | Childhood Cerebral ALD (CCALD): the most severe form, typically presenting in childhood. Adrenomyeloneuropathy (AMN): Onset in adulthood and progresses more slowly. Adrenal Insufficiency Only (AIO): The mildest form may not cause significant neurological symptoms. | Childhood Cerebral ALD (CCALD): Behavior issues Learning difficulties Speech and learning understanding challenges Handwriting deterioration Clumsiness Vision and hearing loss Seizures Swallowing difficulty Dementia Walking challenges Vegetative state Adrenomyeloneuropathy (AMN): Spastic paraparesis Coordination issues Pain Bladder and bowel dysfunction Sexual dysfunction Fatigue Adrenal Insufficiency Only (AIO): Fatigue Muscle weakness Nausea, vomiting, diarrhea Hyperpigmentation Low blood pressure Salty food craving |
| Zellweger Spectrum: includes Zellweger Syndrome (ZS), Neonatal Adrenoleukodystrophy (NALD), Infantile Refsum Disease IRD Sometimes called peroxisome biogenesis disorders (PBDs) A combination of mutations in up to twelve specific genes is involved in Zellweger Spectrum. These involve the assembly of the peroxisome, which breaks down some fats and produces hormones. It affects the brain, liver, and kidneys with resulting difficulty in feeding and movement. | Genetic mutations are present at birth. Differences between types are determined by severity. | Visual difficulties Liver enlargement Kidney issues Cartilage affects Muscle issues Heart and cardiovascular malformation Head malformation |
What are the Symptoms of Leukodystrophy
Leukodystrophy is a group of rare genetic disorders that affect the myelin sheath (nerve protector and insulator) around the nerves. Each type of leukodystrophy is unique to a specific genetic issue. However, symptoms of the disease have commonalities. General leukodystrophy symptoms are listed here. Depending on the affected gene, an individual may have all or just some of these symptoms:
• Cognitive Impairment: Including memory, speech, intellect
• Motor Function Decline: Decreases in motor skills, muscle rigidity, coordination, especially in walking and balance
• Sensory Loss: Issues with vision, hearing, sensation
• Seizures: Focal or generalized
• Behavioral Changes: Irritability, hypersensitivity (especially to the environment)
• Developmental Delays: Prolonged developmental milestone achievement
How is Leukodystrophy Diagnosed
Leukodystrophy can be challenging to diagnose due to the differing symptoms across the many forms. Some cases are undetected. However, tests can diagnose many forms of this rare disease.
Genetic Counseling
If there is a family history of genetic issues such as leukodystrophy, genetic counseling is available to understand your risk of passing a genetic mutation to your child. A blood sample may be taken to establish if either parent has a genetic mutation. Both parents participate in the process. The results of testing will help the parents plan for the future of their child.
Pre-birth testing may include amniocentesis and chorionic villus biopsy for prenatal diagnosis. Both tests are performed by drawing amniotic fluid or placental tissue for genetic testing.
Childhood and Adult Diagnostic Testing
If symptoms appear, your healthcare professional will discuss your family history, perform a physical examination, and a neurological examination to determine your diagnosis.
Blood sample or saliva testing may be done to assess an individual’s genetic code for any mutations that may indicate a form of leukodystrophy.
Imaging studies may be conducted to assess the white matter in your brain and spinal cord. The test most typically used is Magnetic Resonance Imaging (MRI). Diffusion tensor imaging (DTI), an advanced MRI, is another type of imaging study that may be used as an alternative.
A more detailed imaging study is Neurite Orientation Dispersion and Density Imaging (NODDI), which may be needed to detect particular cases of leukodystrophy. In the future, an imaging study called Myelin Water Fraction (MFW) may provide early detection. The MFW quantifies myelin integrity.
Cerebral Spinal Fluid Analysis is a small collection of fluid that surrounds the brain and spinal cord. Cerebral spinal fluid can be withdrawn from the body using a needle inserted into the lower back area between bony vertebrae, into the sac that holds the spinal cord. The spinal cord is not touched in this procedure. A laboratory study is performed on this fluid to look for genetic mutations.
An autopsy performed after death may determine the presence of leukodystrophy and the particular type. This is the only test that will confirm Alexander Disease by the identification of Rosenthal fiber analysis.
What are Tests for Specific Types of Leukodystrophy
Krabbe Disease — dried blood spot assay to assess mutated GALC enzyme activity. Further testing follows if positive. Cerebrospinal fluid will indicate psychosine accumulation, which is indicative of Krabbe disease
Canavan disease is diagnosed from a skin biopsy assessing for skin fibroblasts with NAA (N-acetylaspartate, a brain metabolite). Elevated levels indicate aspartoacylase deficiency (ASPA). The test can separate Canavan disease from other leukodystrophies.
Pelizaeus-Merzbacher Disease (PMD) A molecular study of blood or tissue called Interphase Fluorescence in situ hybridization (FISH) assay, screens for the disease and can detect carriers.
Zellweger syndrome — blood tests for high levels of VLCFA can indicate this genetic mutation form.
What are Treatments for Leukodystrophy
Treatments for leukodystrophies are being researched. Each type of leukodystrophy will require different approaches to assisting with separate genetic issues. Currently, treatments for symptoms of leukodystrophy are available. These include:
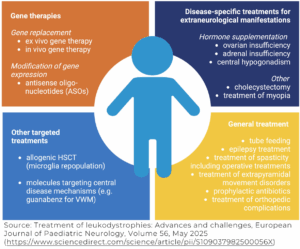
Muscle tone/spasticity: A wide range of treatments is available for muscle tone, also called spasticity. This includes non-pharmaceutical, medication, and Botox injections. A complete listing of treatments is available in the Reeve booklet Managing Spasticity.
Seizures: Various medications are available for seizure control. Different drugs and dosages are selected for individuals’ specific needs, age, and type of seizure.
Dietary adjustment: Altering an individual’s diet can lead to a reduction of over- or underproduction of substances in some cases of leukodystrophy.
Hormone therapy: Can help the functioning of hormonal glands, especially if the adrenal gland function is affected.
Enzyme replacement therapies and substrate reduction therapies: For specific metabolic issues in some forms of leukodystrophy. Additional replacement therapies are being developed.
Bone marrow transplant: Very rarely, this treatment may provide some improvement in select cases of leukodystrophy. More development is needed.
Gene therapy: Changing the way proteins are produced is a goal of gene therapy. Genetic material is used to assist slow or non-functioning genes. This is available only for certain types of leukodystrophy, but it is a goal for researchers.
• Lenmeldy is a gene therapy treatment approved by the Food and Drug Administration (FDA) for individuals with metachromatic leukodystrophy (MLD), which improves an individual’s stem cells.
• Skysona is a gene therapy in the process of FDA approval for boys with early active cerebral ALD (CALD).
• Research is being conducted to develop genetic treatment for individuals with Canavan disease, affecting the nervous system.
Hematopoietic stem cell transplants (HSCT): Replacing diseased stem cells with healthy donor stem cells has been demonstrated to slow some forms of leukodystrophy but not cure it. This treatment is used to correct genetic defects. Recent studies have shown that hematopoietic stem cell transplants have had positive results in children with slowing neurodegeneration in adrenoleukodystrophy, globoid leukodystrophy (Krabbe disease), and metachromatic leukodystrophy.
Bile acids: Chenodeoxycholic acid (CDCA) is a bile acid that may assist individuals with liver dysfunction by dissolving cholesterol gallstones.
Essential fatty acid: In some individuals, docosahexaenoic acid (DHA) improved deficient fatty acids in some individuals with Zellweger syndrome.
To follow the research for improving leukodystrophy conditions, visit the ClinicalTrials.gov. Enter the type of leukodystrophy. Research information about research studies will provide the latest updates.
Rehabilitation or Therapies
Individuals with leukodystrophy will interact with a variety of healthcare professionals. It is wise to select one member of the team, usually the medical specialist in leukodystrophy, to be the leader of the team. This person can assist with sorting through many recommendations to establish the best plan of care for you or your child.
Geneticists can help with future planning and may be involved in genetic treatments.
Specific rehabilitation and therapies depend on the type of leukodystrophy effects. Healthcare providers may include neurologists for seizures, gastrointestinal specialists for digestive issues, and endocrinologists for hormonal issues, among others.
Psychiatrists, psychologists, therapists, and counselors are available for the development of plans to assist with dealing with issues of leukodystrophy to maintain mental wellness of individuals and families.
Physical therapy and occupational therapy professionals provide treatments and therapies for mobility issues, strengthening, positioning, and assistive devices, among others.
Speech and language pathology professionals provide treatments for speaking, swallowing, eating, assistive devices for feeding, and oral motor improvements as needed.
Dieticians and nutritionists can assist with healthy diets that provide the correct calories and energy for individuals of every age. This is especially important in providing the proper nutrition needed, especially if certain foods and elements in foods need to be controlled.
Respiratory therapists can provide exercises for breathing and assistive devices when needed.
Education specialists in the schools can assist children and parents in reaching children’s educational goals. Individual Education Plans (IEPs) are created in conjunction with children and parents to ensure the child’s specific needs are met.
Developmental specialists and child life specialists will help children be children in the adult world of healthcare. They can provide information about reaching developmental milestones.
What is the History of Leukodystrophy
The first cases of leukodystrophy appeared in medical journals in the early 1900s. There were cases prior, but it was not yet identified. The term leukodystrophy originated from Greek words for white (leuko), abnormal (dys), thrive/grow (dystrophy). Together, these words are leukodystrophy, which is how the deteriorating white matter of the brain and spinal cord is now described.
The first type of leukodystrophy described was globoid cell or Krabbe disease, followed by metachromatic leukodystrophy. These early cases were focused on the loss of myelin. Over time, an increased understanding of the genetic code in humans led to the understanding that leukodystrophy is caused by a genetic issue. Now, leukodystrophy is known to affect genes of myelin, glial cells (a specific type of nerve cell), and axons (part of the nerve that transmits signals).
Early diagnosis techniques were less than 50% reliable, with many cases being undiagnosed. As imaging of the nervous system has improved, diagnosis is much clearer.
With the evolution of genetic studies, gene editing, and alteration, the function of genetic mutations has become increasingly understood. More information needs to be gathered to create treatments. Researchers and scientists have been actively working on genetic coding and repair processes to find treatments and eventually cures for leukodystrophies.
Facts and Figures
Each type of leukodystrophy is rare. In the U.S. and Canada, all the leukodystrophy conditions affect anywhere from 1 in 6,000 to 1 in 100,000 live births. In Asian countries, the diseases occur in 3 out of every 100,000 live births. Due to the advances in genetic studies, the occurrence of leukodystrophy may be much higher.
Resources
If you are looking for more information on leukoystrophy or have a specific question, our Information Specialists are available Monday through Friday at 800-539-7309 (toll-free) or online.
Check out our repository of fact sheets on hundreds of topics ranging from state resources to secondary complications of paralysis.
We encourage you to also reach out to leukodystrophy support groups and organizations, including:
United Leukodystrophy Foundation
National Institute of Neurological Disorders and Stroke (NINDS)
National Organization for Rare Diseases (NORD)
Searching for a specific leukodystrophy will also provide organizations and foundations that support that particular type.
Clinical guidelines:
Adult-Onset Leukodsytrophies: A Practical Guide, Recent Treatment Updates, and Future Directions via PubMed Central Frontiers in Neurology 2023 Jul 26;14:1219324.
Clinical Practice Guidelines for the Diagnosis, Management, and Surveillance of LMNB1-Related Autosomal Dominant Leukodystrophy Neurology Genetics October 2025
Consensus Guidelines for the Monitoring and Management of Metachromatic Leukodystrophy in the United States: https://pubmed.ncbi.nlm.nih.gov/38613540/ via PubMed Cytotherapy 2024 Jul:26(7):739-748. Doi: 10.1016/j.jcyt.2024.03.487. Epub2024 Apr 1.
Hematopoietic Stem Cell Transplants:
Page KM, Stenger EO, Connelly JA, et.al. Hematopoietic Stem Cell Transplantation to Treat Leukodystrophies: Clinical Practice Guidelines from the Hunter’s Hope Leukodystrophy Care Network. Biol Blood Marrow Transplant. 2019 Dec;25(12):e363-e374. doi: 10.1016/j.bbmt.2019.09.003.
Epub 2019 Sep 6. PMID: 31499213.
References
Adang L. Leukodystrophies. Continuum (Minneap Minn). 2022 Aug 1;28(4):1194-1216. doi: 10.1212/CON.0000000000001130. PMID: 35938662; PMCID: PMC11320896.
Aerts-Kaya F, van Til NP. Gene and Cellular Therapies for Leukodystrophies. Pharmaceutics. 2023 Oct 24;15(11):2522. doi: 10.3390/pharmaceutics15112522. PMID: 38004502; PMCID: PMC10675548.
Ashrafi MR, Amanat M, Garshasbi M, Kameli R, Nilipour Y, Heidari M, Rezaei Z, Tavasoli AR. An Update on Clinical, Pathological, Diagnostic, and Therapeutic Perspectives of Childhood Leukodystrophies. Expert Rev Neurother. 2020 Jan;20(1):65-84. doi: 10.1080/14737175.2020.1699060. Epub 2019 Dec 12. PMID: 31829048.
Axelsen TM, Vammen TL, Bak M, Pourhadi N, Stenør CM, Grønborg S. Case Report: ‘AARS2 Leukodystrophy’. Mol Genet Metab Rep. 2021 Jul 13;28:100782. doi: 10.1016/j.ymgmr.2021.100782. PMID: 34285876; PMCID: PMC8280508.
Belleri M, Presta M. Endothelial Cell Dysfunction in Globoid Cell Leukodystrophy. J Neurosci Res. 2016 Nov;94(11):1359-67. doi: 10.1002/jnr.23744. Epub 2016 Apr 1. PMID: 27037626.
Burlina AP, Manara R, Gueraldi D. Lysosomal Storage Diseases. Handb Clin Neurol. 2024;204:147-172. doi: 10.1016/B978-0-323-99209-1.00008-9. PMID: 39322377.
Ceravolo G, Zhelcheska K, Squadrito V, Pellerin D, Gitto E, Hartley L, Houlden H. Update on Leukodystrophies and Developing Trials. J Neurol. 2024 Jan;271(1):593-605. doi: 10.1007/s00415-023-11996-5. Epub 2023 Sep 27. PMID: 37755460; PMCID: PMC10770198.
Cheon JE, Kim IO, Hwang YS, Kim KJ, Wang KC, Cho BK, Chi JG, Kim CJ, Kim WS, Yeon KM. Leukodystrophy in Children: A Pictorial Review of MR Imaging Features. Radiographics. 2002 May-Jun;22(3):461-76. doi: 10.1148/radiographics.22.3.g02ma01461. PMID: 12006681.
Coulombe B, Derksen A, La Piana R, Brais B, Gauthier MS, Bernard G. POLR3-Related Leukodystrophy: How Do Mutations Affecting RNA Polymerase III Subunits Cause Hypomyelination? Fac Rev. 2021 Feb 5;10:12. doi: 10.12703/r/10-12. PMID: 33659930; PMCID: PMC7894263.
do Rosario MC, Bey GR, Nmezi B, Liu F, Oranburg T, Cohen ASA, Coffman KA, Brown MR, Kiselyov K, Waisfisz Q, Flohil MT, Siddiqui S, Rosenfeld JA, Iglesias A, Girisha KM, Wolf NI, Padiath QS, Shukla A. Variants in the Zinc Transporter TMEM163 Cause a Hypomyelinating Leukodystrophy. Brain. 2022 Dec 19;145(12):4202-4209. doi: 10.1093/brain/awac295. PMID: 35953447; PMCID: PMC10200305.
Engelen M, van der Knaap MS, Wolf NI. Amino-acyl tRNA Synthetases Associated with Leukodystrophy. Handb Clin Neurol. 2024;204:253-261. doi: 10.1016/B978-0-323-99209-1.00020-X. PMID: 39322382.
Garcia LM, Hacker JL, Sase S, Adang L, Almad A. Glial Cells in the Driver Seat of Leukodystrophy Pathogenesis. Neurobiol Dis. 2020 Dec;146:105087. doi: 10.1016/j.nbd.2020.105087. Epub 2020 Sep 23. PMID: 32977022.
Gavazzi F, Charsar B, Hamilton E, Erler JA, Patel V, Woidill S, Sevagamoorthy A, Helman G, Schmidt J, Pizzino A, Muirhead K, Takanohashi A, Bonkowsky JL, Meyerhoffer K, Simons C, Doi H, Satoko M, Matsumoto N, Delgado MR, Sanchez-Castillo M, Wang J, de Carvalho DR, Tournev I, Chamova T, Jordanova A, Clegg NJ, Nicita F, Bertini E, Teng M, Williams D, Tonduti D, Houlden H, Stellingwerff M, Wassmer E, Garcia-Cazorla A, Bernard G, Mirchi A, Toutounchi H, Wolf NI, van der Knaap MS, Shults J, Adang LA, Vanderver AL. The Natural History of Variable Subtypes in Pediatric-Onset TUBB4A-Related Leukodystrophy. Mol Genet Metab. 2025 Mar;144(3):109048. doi: 10.1016/j.ymgme.2025.109048. Epub 2025 Feb 1. PMID: 39951964; PMCID: PMC11875891.
Helman G, Van Haren K, Bonkowsky JL, Bernard G, Pizzino A, Braverman N, Suhr D, Patterson MC, Ali Fatemi S, Leonard J, van der Knaap MS, Back SA, Damiani S, Goldman SA, Takanohashi A, Petryniak M, Rowitch D, Messing A, Wrabetz L, Schiffmann R, Eichler F, Escolar ML, Vanderver A; GLIA Consortium. Disease Specific Therapies in Leukodystrophies and Leukoencephalopathies. Mol Genet Metab. 2015 Apr;114(4):527-36. doi: 10.1016/j.ymgme.2015.01.014. Epub 2015 Feb 7. PMID: 25684057; PMCID: PMC4390468.
Hol EM, Dykstra W, Chevalier J, Cuadrado E, Bugiani M, Aronica E, Verkhratsky A. Neuroglia in Leukodystrophies. Handb Clin Neurol. 2025;210:159-175. doi: 10.1016/B978-0-443-19102-2.00032-6. PMID: 40148043.
Hooshmand SJ, Chohan KL, Raghunathan A, Renaud DL, Ruff MW. BRAT1-Associated Leukodystrophy Exacerbated by Classic Hodgkin Lymphoma-Directed Therapy. Neurologist. 2024 May 1;29(3):170-172. doi: 10.1097/NRL.0000000000000539. PMID: 38019165.
Jaunmuktane Z. Neuropathology of White Matter Disorders. Handb Clin Neurol. 2024;204:3-20. doi: 10.1016/B978-0-323-99209-1.00011-9. PMID: 39322386.
Jorge MS, Bugiani M. Astroglia in Leukodystrophies. Adv Exp Med Biol. 2019;1175:199-225. doi: 10.1007/978-981-13-9913-8_9. PMID: 31583590.
Kemec Z, Tüzün C, Gürel A. Neurogenic Bladder and Acute Kidney Injury in Leukodystrophy. Cureus. 2020 Jun 20;12(6):e8707. doi: 10.7759/cureus.8707. PMID: 32699703; PMCID: PMC7372234.
Khalaf G, Mattern C, Begou M, Boespflug-Tanguy O, Massaad C, Massaad-Massade L. Mutation of Proteolipid Protein 1 Gene: From Severe Hypomyelinating Leukodystrophy to Inherited Spastic Paraplegia. Biomedicines. 2022 Jul 15;10(7):1709. doi: 10.3390/biomedicines10071709. PMID: 35885014; PMCID: PMC9313024.
Köhler W, Curiel J, Vanderver A. Adulthood Leukodystrophies. Nat Rev Neurol. 2018 Feb;14(2):94-105. doi: 10.1038/nrneurol.2017.175. Epub 2018 Jan 5. PMID: 29302065; PMCID: PMC11348681.
Koto Y, Ueki S, Yamakawa M, Sakai N. Experiences of Patients with Metachromatic Leukodystrophy, Adrenoleukodystrophy, or Krabbe Disease and the Experiences of Their Family Members: A Qualitative Systematic Review. JBI Evid Synth. 2024 Jul 1;22(7):1262-1302. doi: 10.11124/JBIES-23-00303. PMID: 38533650; PMCID: PMC11230659.
Kuhn J, Cascella M. Alexander Disease. 2023 Sep 4. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 32965913.
Lamichhane A, Rocha Cabrero F. Metachromatic Leukodystrophy. 2023 Jul 17. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 32809579.
Lanciotti A, Brignone MS, Bertini E, Petrucci TC, Aloisi F, Ambrosini E. Astrocytes: Emerging Stars in Leukodystrophy Pathogenesis. Transl Neurosci. 2013 Jun 1;4(2):10.2478/s13380-013-0118-1. doi: 10.2478/s13380-013-0118-1. PMID: 24340223; PMCID: PMC3856885.
Lin DS, Ho CS, Huang YW, Wu TY, Lee TH, Huang ZD, Wang TJ, Yang SJ, Chiang MF. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells. 2020 May 1;9(5):1124. doi: 10.3390/cells9051124. PMID: 32370022; PMCID: PMC7290671.
Lin DS, Huang YW, Lee TH, Chang L, Huang ZD, Wu TY, Wang TJ, Ho CS. Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy. Cells. 2023 Mar 24;12(7):993. doi: 10.3390/cells12070993. PMID: 37048066; PMCID: PMC10093124.
Modesti NB, Evans SH, Jaffe N, Vanderver A, Gavazzi F. Early Recognition of Patients with Leukodystrophies. Curr Probl Pediatr Adolesc Health Care. 2022 Dec;52(12):101311. doi: 10.1016/j.cppeds.2022.101311. Epub 2022 Dec 2. PMID: 36470810; PMCID: PMC11326772.
Murofushi Y, Hayakawa I, Abe Y, Ohto T, Murayama K, Suzuki H, Takenouchi T, Kosaki K, Kubota M. Ketogenic Diet for KARS-Related Mitochondrial Dysfunction and Progressive Leukodystrophy. Neuropediatrics. 2022 Feb;53(1):65-68. doi: 10.1055/s-0041-1732446. Epub 2021 Aug 26. PMID: 34448181.
Nowacki JC, Fields AM, Fu MM. Emerging Cellular Themes in Leukodystrophies. Front Cell Dev Biol. 2022 Aug 8;10:902261. doi: 10.3389/fcell.2022.902261. PMID: 36003149; PMCID: PMC9393611.
Page KM, Stenger EO, Connelly JA, Shyr D, West T, Wood S, Case L, Kester M, Shim S, Hammond L, Hammond M, Webb C, Biffi A, Bambach B, Fatemi A, Kurtzberg J. Hematopoietic Stem Cell Transplantation to Treat Leukodystrophies: Clinical Practice Guidelines from the Hunter’s Hope Leukodystrophy Care Network. Biol Blood Marrow Transplant. 2019 Dec;25(12):e363-e374. doi: 10.1016/j.bbmt.2019.09.003. Epub 2019 Sep 6. PMID: 31499213.
Perlman SJ, Mar S. Leukodystrophies. Adv Exp Med Biol. 2012;724:154-71. doi: 10.1007/978-1-4614-0653-2_13. PMID: 22411242.
Perrier S, Michell-Robinson MA, Bernard G. POLR3-Related Leukodystrophy: Exploring Potential Therapeutic Approaches. Front Cell Neurosci. 2021 Jan 28;14:631802. doi: 10.3389/fncel.2020.631802. PMID: 33633543; PMCID: PMC7902007.
Sevin C, Mochel F. Hematopoietic Stem Cell Transplantation in Leukodystrophies. Handb Clin Neurol. 2024;204:355-366. doi: 10.1016/B978-0-323-99209-1.00017-X. PMID: 39322389.
Singh S, Mishra A, Murthy C, Inban P, Abdefatah Ali M, Yadav AS, Intsiful TA, O Omar ZT, Lakhra S, Khan DA. A Rare Case of Hypomyelinating Leukodystrophy and Its Management: A Case Report and Literature Review. Cureus. 2023 Mar 21;15(3):e36471. doi: 10.7759/cureus.36471. PMID: 37090362; PMCID: PMC10117409.
Souza PVS, Badia BML, Silva LHL, Teixeira CAC, Seneor DD, Marin VDGB, Farias IB, Dias RB, Oliveira ASB, Pinto WBVR. Leukodystrophy with Disorders of Sex Development Due to WT1 Mutations. J Neurol Sci. 2018 Jul 15;390:94-98. doi: 10.1016/j.jns.2018.04.020. Epub 2018 Apr 13. PMID: 29801916.
Tobias JD. Anaesthetic Considerations for the Child with Leukodystrophy. Can J Anaesth. 1992 Apr;39(4):394-7. doi: 10.1007/BF03009053. PMID: 1563064.
Raymond GV. Leukodystrophy: Basic and Clinical. Adv Neurobiol. 2017;15:365-382. doi: 10.1007/978-3-319-57193-5_14. PMID: 28674989.
Resende LL, de Paiva ARB, Kok F, da Costa Leite C, Lucato LT. Adult Leukodystrophies: A Step-by-Step Diagnostic Approach. Radiographics. 2019 Jan-Feb;39(1):153-168. doi: 10.1148/rg.2019180081. PMID: 30620693.
Rosenberg JB, Kaminsky SM, Aubourg P, Crystal RG, Sondhi D. Gene Therapy for Metachromatic Leukodystrophy. J Neurosci Res. 2016 Nov;94(11):1169-79. doi: 10.1002/jnr.23792. PMID: 27638601; PMCID: PMC5027970.
Rutherford HA, Hamilton N. Animal Models of Leukodystrophy: A New Perspective for the Development of Therapies. FEBS J. 2019 Nov;286(21):4176-4191. doi: 10.1111/febs.15060. Epub 2019 Oct 9. PMID: 31520449.
Sferra A, Fortugno P, Motta M, Aiello C, Petrini S, Ciolfi A, Cipressa F, Moroni I, Leuzzi V, Pieroni L, Marini F, Boespflug Tanguy O, Eymard-Pierre E, Danti FR, Compagnucci C, Zambruno G, Brusco A, Santorelli FM, Chiapparini L, Francalanci P, Loizzo AL, Tartaglia M, Cestra G, Bertini E. Biallelic Mutations in RNF220 Cause Laminopathies Featuring Leukodystrophy, Ataxia and Deafness. Brain. 2021 Nov 29;144(10):3020-3035. doi: 10.1093/brain/awab185. PMID: 33964137.
Soderholm HE, Chapin AB, Bayrak-Toydemir P, Bonkowsky JL. Elevated Leukodystrophy Incidence Predicted from Genomics Databases. Pediatr Neurol. 2020 Oct;111:66-69. doi: 10.1016/j.pediatrneurol.2020.06.005. Epub 2020 Jun 17. PMID: 32951664; PMCID: PMC7506144.
Suzuki K. Globoid Cell Leukodystrophy (Krabbe’s Disease): Update. J Child Neurol. 2003 Sep;18(9):595-603. doi: 10.1177/08830738030180090201. PMID: 14572137.
Takanashi JI. Magnetic Resonance Imaging and Spectroscopy in Hypomyelinating Leukodystrophy. Brain Dev. 2025 Jun;47(3):104345. doi: 10.1016/j.braindev.2025.104345. Epub 2025 Apr 1. PMID: 40174481.
Thakkar RN, Patel D, Kioutchoukova IP, Al-Bahou R, Reddy P, Foster DT, Lucke-Wold B. Leukodystrophy Imaging: Insights for Diagnostic Dilemmas. Med Sci (Basel). 2024 Jan 25;12(1):7. doi: 10.3390/medsci12010007. PMID: 38390857; PMCID: PMC10885080.
Tonduti D, Zambon AA, Ghezzi D, Lamantea E, Izzo R, Parazzini C, Baldoli C, van der Knaap MS, Fumagalli F. Expanding the Spectrum of NUBPL-Related Leukodystrophy. Neuropediatrics. 2023 Jun;54(3):161-166. doi: 10.1055/s-0043-1764214. Epub 2023 Mar 3. PMID: 36868263.
van der Knaap MS, Bugiani M. Leukodystrophies: A Proposed Classification System Based on Pathological Changes and Pathogenetic Mechanisms. Acta Neuropathol. 2017 Sep;134(3):351-382. doi: 10.1007/s00401-017-1739-1. Epub 2017 Jun 21. PMID: 28638987; PMCID: PMC5563342.
Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, Garbern J, Eichler F, Labauge P, Aubourg P, Rodriguez D, Patterson MC, Van Hove JL, Schmidt J, Wolf NI, Boespflug-Tanguy O, Schiffmann R, van der Knaap MS; GLIA Consortium. Case Definition and Classification of Leukodystrophies and Leukoencephalopathies. Mol Genet Metab. 2015 Apr;114(4):494-500. doi: 10.1016/j.ymgme.2015.01.006. Epub 2015 Jan 29. PMID: 25649058; PMCID: PMC4390457.
Waldman AT. Leukodystrophies. Continuum (Minneap Minn). 2018 Feb;24(1, Child Neurology):130-149. doi: 10.1212/CON.0000000000000560. PMID: 29432240.
Wolf NI, Engelen M, van der Knaap MS. MRI Pattern Recognition in White Matter Disease. Handb Clin Neurol. 2024;204:37-50. doi: 10.1016/B978-0-323-99209-1.00019-3. PMID: 39322391.
Wolf NI, van der Knaap MS, Engelen M. Treatment of Leukodystrophies: Advances and Challenges. Eur J Paediatr Neurol. 2025 May;56:46-50. doi: 10.1016/j.ejpn.2025.03.016. Epub 2025 Apr 15. PMID: 40279833.
Wolf NI, Ffrench-Constant C, van der Knaap MS. Hypomyelinating Leukodystrophies – Unravelling Myelin Biology. Nat Rev Neurol. 2021 Feb;17(2):88-103. doi: 10.1038/s41582-020-00432-1. Epub 2020 Dec 15. PMID: 33324001.
Zhang T, Yan C, Liu Y, Cao L, Ji K, Li D, Chi L, Zhao Y. Late-Onset Leukodystrophy Mimicking Hereditary Spastic Paraplegia without Diffuse Leukodystrophy on Neuroimaging. Neuropsychiatr Dis Treat. 2021 May 12;17:1451-1458. doi: 10.2147/NDT.S296424. PMID: 34012265; PMCID: PMC8126967.
