Amyotrophie spinale
L’atrofia muscolare spinale, più comunemente nota come SMA, è un gruppo di malattie genetiche che colpisce sia il sistema nervoso centrale (SNC), ovvero il cervello e il midollo spinale, sia il sistema nervoso periferico (SNP), che comprende tutti gli altri nervi del corpo. Solitamente, la malattia progredisce con una crescente debolezza muscolare, man mano che il bambino invecchia. La debolezza e l’atrofia (deperimento muscolare) sono più pronunciate verso il centro del corpo rispetto alle estremità. La SMA è solitamente simmetrica su entrambi i lati del corpo.
Nella SMA sono colpiti in particolare i nervi motori del midollo spinale nel corno anteriore (la materia grigia accanto al canale centrale). Questo interrompe i messaggi inviati dal cervello al corpo. Il messaggio di movimento non può essere eseguito attraverso l’uso dei muscoli scheletrici al fine di creare volontariamente quel movimento. Poiché i muscoli del corpo non ricevono i messaggi dai nervi, si atrofizzano e si riducono nel tempo. Particolarmente colpiti sono i movimenti di viso, lingua, gola, braccia, petto e gambe. Ciò influisce su funzioni quali la respirazione, la deglutizione, il linguaggio e la deambulazione.

Le funzioni corporee che non sono colpite dalla SMA includono funzioni mentali come l’apprendimento e le abilità sociali. I nervi sensoriali non sono colpiti, per cui le sensazioni o le percezioni del corpo si mantengono intatte. La vista e l’udito non sono interessati. Le funzioni intestinali e vescicali rimangono inalterate.
Genetica
La SMA è comunemente causata da una mutazione o da un gene mancante, il che la rende una malattia genetica (ereditaria). La SMN è una proteina necessaria per la sopravvivenza dell’organismo, poiché una sua carenza provoca la perdita selettiva dei motoneuroni. Il gene affetto che causa la SMA è il gene 1 del motoneurone di sopravvivenza (SMN1), situato sul cromosoma 5q. Questo gene è responsabile per il mantenimento della salute e della funzione dei motoneuroni, che sono i nervi che causano il movimento nel corpo. Il gene SMN2 ordina all’organismo di produrre il gene SMN1. Se SMN1 è assente e SMN2 è assente, la proteina SMN non può essere prodotta. I soggetti privi del gene SMN1 sono affetti da SMA, anche se alcuni individui privi di SMN1 possono contare sul gene SMN2 per produrre la proteina SMN.
Per avere la SMA, le persone devono ereditare un gene recessivo (non dominante) da entrambi i genitori. Ereditando un solo gene recessivo da un genitore non si contrae la malattia, ma l’individuo è un portatore sano della SMA. In alcuni rari casi i fattori genetici non sono chiari.
Ci sono molte domande sull’aspettativa di vita con la SMA. Tradizionalmente, per il tipo di SMA più basso l’aspettativa di vita è inferiore. Il tipo IV, il più elevato, ha un’aspettativa di vita pari a quella della popolazione generale. Tuttavia, grazie allo sviluppo di nuovi trattamenti per migliorare la SMA, le aspettative di vita stanno aumentando. Inoltre, i ricercatori stanno incrementando costantemente le opzioni volte a migliorare la respirazione, la mobilità e le altre complicazioni della SMA.
Tipi di SMA
Esistono cinque tipi di SMA che sono basati sulla migliore funzione motoria. Nel 1991 l’Associazione per la distrofia muscolare (MDA – Muscular Dystrophy Association) ha elaborato un sistema di classificazione della SMA. Il grado di gravità varia all’interno dei tipi. I tipi possono essere suddivisi in più categorie. Solo il 25% circa delle persone affette da SMA può essere chiaramente classificato entro un unico tipo. Probabilmente avete già sentito nominare questo sistema di classificazione “a tipi”.
Sistema di classificazione originale della SMA
| Tipo di SMA | Età di diagnosi/insorgenza | Segni clinici | Capacità |
| SMA di tipo 0 | Prenatale | – Diminuzione dei movimenti fetali – Riflessi scarsi o assenti – Paralisi facciale – Difetto del setto cardiaco – Contratture articolari e problemi scheletrici – Contrazioni muscolari – Difficoltà di deglutizione e alimentazione – Insufficienza respiratoria | È necessario un supporto per la respirazione |
| SMA di tipo I detta anche malattia di Werdnig-Hoffmann | 0-6 mesi | – Basso tono corporeo (postura “a rana”) – Scarso controllo della testa – Riflessi ridotti o non reattivi – Respirazione addominale dovuta alla forza limitata dei muscoli intercostali (quelli tra le costole), ma diaframma funzionante – Rischio di aspirazione a causa della debolezza della motricità orale – Processi mentali non alterati | Incapacità di rotolare o sedersi autonomamente |
| SMA di tipo II | Meno di 18 mesi | – In genere, debolezza progressiva nelle gambe, ma non altrettanto nelle braccia – Tono basso, senza riflessi – Complicazioni ossee dovute alla debolezza dei muscoli, come scoliosi, contratture, irrigidimento della mandibola – Malattia polmonare di tipo restrittivo – Processi mentali non alterati | Capacità di stare seduti ma non di camminare/stare in piedi |
| SMA di tipo III detta anche malattia di Kugelberg-Welander | Più di 18 mesi Sottotipi: 3a 18 mesi – 3 anni 3b >3 anni | – Debolezza delle gambe superiore a quella delle braccia – Debolezza dei muscoli respiratori o scoliosi lieve o nulla – Difficoltà a correre, salire le scale, tentare di stare in piedi – Processi mentali non alterati – Aspettativa di vita inalterata | Cammina ma potrebbe iniziare in ritardo |
| SMA di tipo IV | Più di 21 anni o in età adulta | – 5% dei casi – Insorgenza in età adulta, spesso intorno ai 30 anni ma può essere più precoce o più tardiva – Debolezza delle gambe da lieve a moderata – Processi mentali non alterati – Aspettativa di vita inalterata | Cammina in modo indipendente o con dispositivi di assistenza alla mobilità |
| SMA non legata al cromosoma 5 (SMN1 o proteina SMN) | Variabile | – Gravità variabile – Può includere i muscoli più distanti dal centro del corpo | Variabili |
Sistema di classificazione attuale della SMA
La SMA è un problema di salute che cambia nel corso del tempo. Alcune persone saranno classificate come sopra, mentre altre passeranno da un tipo all’altro in base alla loro capacità funzionale. Il passaggio può avvenire sulla base di guadagni o perdite di funzionalità a seconda dell’individuo. Pertanto, è stato sviluppato un sistema di classificazione più pratico che indica il livello di funzionalità attuale, piuttosto che quello presente al momento della diagnosi. Questo sistema di classificazione è suddiviso in tre categorie, a cui si aggiungono delle sotto-categorie man mano che vengono condotte ulteriori ricerche. Le classificazioni sono Nonsitters, Sitters e Walkers. Nessuno rientra perfettamente in una categoria e i cambiamenti di categoria sono frequenti.
| Categoria | Classificazione per tipo | Caratteristiche |
| Nonsitters | Principalmente di tipo I | – Difficoltà di respirazione e di alimentazione – Deformità del torace a campana – Fascicolazioni nella lingua – Assenza di flessione dell’anca, testa all’indietro (head lag) |
| Sitters | Principalmente di tipo II e III | – Riesce a stare seduto ma non a camminare – La debolezza muscolare è più intensa nelle gambe che nelle braccia – Sviluppo di contratture articolari, anchilosi mandibolare e spesso scoliosi – La maggior parte non presenta problemi di respirazione e deglutizione; tuttavia, la debolezza dei muscoli intercostali può portare a una malattia polmonare di tipo restrittivo e a un’insufficienza respiratoria – La SMA può progredire tra i 5 e i 15 anni, soprattutto durante la pubertà |
| Walkers | Tipo II o IV | – Individui che raggiungono o mantengono la capacità di camminare – Minore funzionalità delle gambe rispetto alla debolezza delle braccia – In genere, nessuna difficoltà di respirazione o di deglutizione, tuttavia, nel corso della vita, la deambulazione e la respirazione possono diminuire molto lentamente e leggermente – Il 95% dei bambini che hanno iniziato a sedersi precocemente (sitters precoci) può camminare entro i 18 mesi di età – Il 50% dei bambini che hanno iniziato a sedersi tardivamente (sitters tardivi) può camminare entro i 18 mesi – L’aspettativa di vita è pari a quella della popolazione generale |
| Insorgenza negli adulti | Tipo IV | – I sintomi possono manifestarsi con fastidio o dolore ai muscoli delle gambe – Le gambe possono diventare deboli e richiedere l’uso di stecche o tutori per mantenere la deambulazione – La deglutizione e la respirazione non sono in genere influenzate |
Sintomi/Diagnosi
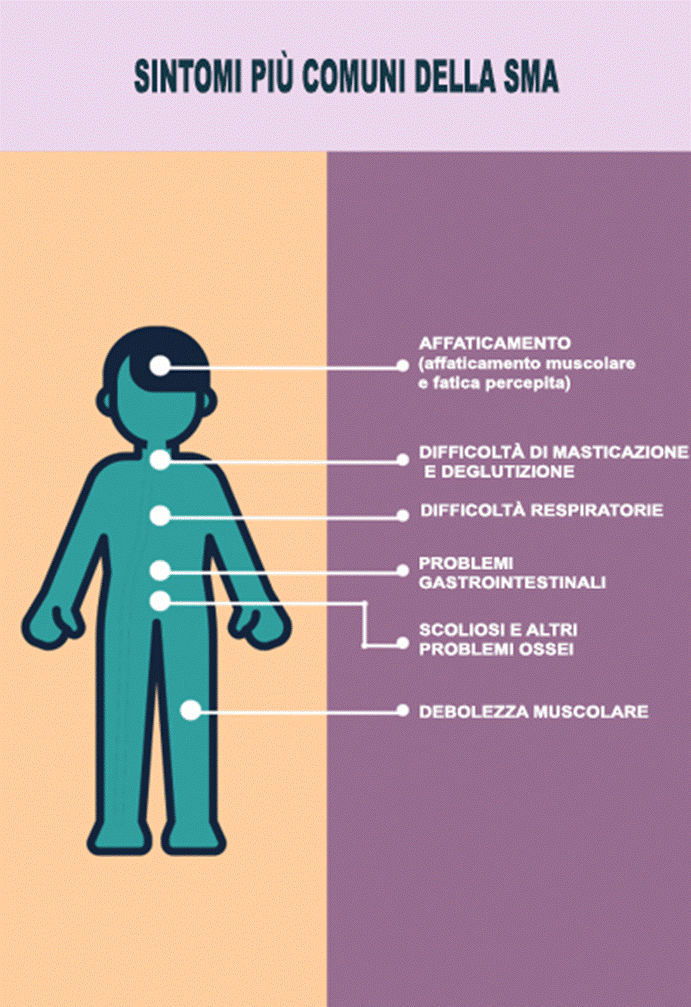
Generalmente, la debolezza muscolare è il primo sintomo della SMA. Se la debolezza muscolare è grave, in particolare con una maggiore debolezza verso il centro del corpo e minore nelle estremità, la diagnosi sarà abbastanza chiara. Debolezze lievi potrebbero non portare direttamente alla diagnosi di SMA.
Il test genetico che utilizza un campione di sangue per valutare la presenza di mutazioni o delezioni dei geni SMN1 e SMN2 è il test definitivo. Inoltre, tale test è in grado di individuare i portatori sani di SMA. Se un uomo e una donna sono entrambi portatori sani, possono decidere di ricorrere a un donatore per ridurre il rischio di malattia nel bambino. Il test genetico per la SMA è affidabile al 95%.
L’elettromiografia (EMG) o test della funzione muscolare mediante la rilevazione dell’attività elettrica dei muscoli può essere eseguita confrontando la contrazione e il rilassamento muscolare.
Gli studi di conduzione nervosa (NCS) possono essere eseguiti al momento dell’EMG. Questi test valutano la capacità di un nervo di trasmettere messaggi.
Questo grafico è stato ristampato con il permesso di SMA News Today.
Con una biopsia muscolare, un frammento estremamente piccolo di un muscolo viene rimosso dal corpo ed esaminato al microscopio per individuare eventuali cambiamenti che indichino una malattia neuromuscolare.
Biomarcatori Le variazioni di alcune caratteristiche del corpo o del sangue, note come biomarcatori, possono indicare la presenza di malattie. Il numero di copie di SMN2 è inversamente correlato alla diagnosi di SMA e ne indica la gravità. Le misurazioni tramite EMG del complesso dei potenziali d’azione muscolari, la stima delle unità motorie e la miografia a impedenza elettrica sono esami predittivi della SMA e permettono di stimare i farmaci necessari per il trattamento.
Altri esami del sangue, tra cui i livelli di mRNA e proteina SMN, possono rivelare i materiali di scarto della distruzione muscolare.
Trattamento
Sono disponibili farmaci per dei tipi specifici di SMA.
Il Nusinersen (SPINRAZA) aumenta la produzione della proteina SMN attraverso la modulazione dello splicing SMN2 che mantiene i motoneuroni. Questo farmaco viene somministrato per via intratecale (nello spazio tra il midollo spinale e la membrana che lo contiene) con un’iniezione spinale. Disponibile per bambini e adulti con qualsiasi tipo di SMA.
L’Onasemnogene abeparvovec-xioi (Zolgensma) è una terapia di sostituzione del gene SMN1. La produzione della proteina SMN viene migliorata mediante il trasferimento del gene SMN1 nelle cellule neurali (nervose) utilizzando virus adeno-associati. Questi virus non sono patogeni (non fanno ammalare il bambino). Passano facilmente nelle cellule e sono comunemente utilizzati nella terapia genica grazie alla loro sicurezza. Ulteriori informazioni sono contenute nell’articolo di Pillay, 2017, nella sezione di riferimento. Il farmaco è disponibile per i bambini di età inferiore ai due anni con SMA a esordio infantile. L’obiettivo è migliorare il movimento, la funzione e la sopravvivenza dei muscoli.
Il Risdiplam (Evrysdi) è un farmaco orale che stimola l’espressione del gene SMN3 della proteina SMN. È indicato per i bambini a partire dai due mesi di vita.
Da quando è stato scoperto il gene SMN, abbiamo avuto un numero molto alto di studi sui trattamenti per la SMA. Ciò ha prodotto studi di laboratorio e studi clinici (su esseri umani) che sono in corso. Questi studi includono la terapia a piccole molecole per aumentare la proteina SMN, la terapia a base di RNA per aumentare la SMN2 nella molecola di RNA e causare una maggiore espressione della SMN2 e la terapia genica per sostituire il gene SMN1 mancante.
Altri studi sono in corso di realizzazione. Si consiglia di consultare il sito web: https://clinicaltrials.gov/ per i nuovi studi clinici sponsorizzati dal governo degli Stati Uniti. Le informazioni sui singoli studi clinici per la SMA vi consentiranno di conoscere le idee dei ricercatori e le ultime informazioni sugli studi. È importante sapere cosa succederà, in modo da potersi preparare e prendere delle decisioni ben informate sulla salute di vostro figlio.
Terapie riabilitative
Un team di professionisti sanitari si prenderà cura del bambino. Ogni professionista porta la propria specializzazione per soddisfare le esigenze individuali, lavorando all’unisono.
La diagnosi viene fatta da un genetista o da un neurologo, che vengono consultati nel caso in cui si sospetti la SMA. Può essere un’esperienza emotivamente logorante. Avere informazioni sulla malattia è utile per capire quali opzioni di trattamento medico saranno disponibili per vostro figlio e come mantenere la sua qualità di vita e le sue esperienze. Questa persona contribuirà a definire il Programma o Piano Educativo Individualizzato (PEI) che sarà necessario a scuola per gestire le esigenze specifiche di vostro figlio. Può anche aiutare ulteriormente con la pianificazione familiare. Sarà in grado di spiegarvi le opzioni per le terapie in fase di ricerca se siete interessati a seguire tali percorsi di cura.
Il benessere mentale può essere migliorato da uno psicologo che lavora con i bambini e con le famiglie. Questa persona è utile per gestire le emozioni e gli effetti della malattia su vostro figlio, sulla vostra famiglia e su di voi. Questa persona contribuirà anche a definire il Programma Educativo Individualizzato (PEI) che sarà necessario a scuola per gestire le esigenze specifiche di vostro figlio.
L’assistenza sanitaria specialistica sarà fornita da uno specialista in disturbi neuromuscolari pediatrici, da un fisiatra pediatrico o da un fisiatra per adulti (in base all’età). Questo medico si occuperà delle esigenze funzionali del bambino sia per favorire le capacità (apprendimento di nuove abilità) sia per la riabilitazione (riapprendimento di abilità). Tale medico può guidare l’equipe sanitaria nell’organizzazione e nella fornitura di cure mediche, attrezzature, terapie, farmaci e test. Conosce sia le ricerche sia i nuovi trattamenti che potrebbero essere utili.
Agli educatori scolastici, come ad esempio il dirigente scolastico, gli insegnanti e gli assistenti didattici si potrà richiedere un supporto all’apprendimento per il bambino. Il coinvolgimento del personale scolastico attraverso l’uso del PEI è essenziale. Con la SMA, la capacità di apprendimento non è compromessa, ma potrebbero essere richieste attrezzature adattive per respirazione e dispositivi di mobilità, per prendere appunti, per l’attività fisica, per l’assistenza all’alimentazione, per periodi riposo e per altre questioni. Il PEI è un piano creato e documentato in base alle esigenze di vostro figlio che descrive il modo in cui il sistema scolastico soddisferà tali necessità.
Le attrezzature necessarie per ottenere la massima indipendenza saranno consigliate da fisioterapisti, ergoterapisti e logopedisti. Le attrezzature per la postura sono necessarie per mantenere l’allineamento del corpo e sostenere la respirazione, il sistema scheletrico la digestione e molte altre questioni. Le attrezzature possono includere sistemi di riposo, sedute, sedie a rotelle, deambulatori, tutori e attrezzature per il bagno.
Le terapie per l’atrofia muscolare spinale sono adattate alle esigenze individuali. Non tutti gli individui avranno bisogno di tutti questi trattamenti, ma sapere che sono disponibili è utile per migliorare la salute e per evitare problemi di salute.
Respirazione I trattamenti respiratori necessari sono prescritti e coordinati da uno pneumologo e da un terapista respiratorio pediatrico o dell’adulto. Questi trattamenti possono includere la ventilazione meccanica che potrebbe richiedere una tracheostomia, la ventilazione meccanica non invasiva che non richiede una tracheostomia, l’aspirazione, l’insufflazione, i trattamenti con nebulizzatori ed esercizi di respirazione come la spirometria incentivante. Le infezioni respiratorie possono insorgere più facilmente, pertanto il lavaggio delle mani, gli esercizi di respirazione, il distanziamento e l’isolamento dei soggetti malati sono precauzioni importanti.
Alcuni bambini possono soffrire di apnea notturna quando sono a riposo. Assicurarsi di fare una valutazione e di monitorare i sintomi come russare, rantolare, periodi di assenza di respiro o mal di testa mattutino quando è sdraiato. I problemi di respirazione possono causare altre condizioni di salute e colpire la funzione cardiaca.
Le abilità motorie orali possono essere rafforzate e migliorate con l’assistenza di un logopedista per aiutare il bambino a masticare, deglutire e migliorare l’articolazione del linguaggio. Se la deglutizione è un problema, si possono utilizzare metodi di alimentazione alternativi, come ad esempio un sondino.
Comunicazione I problemi di motricità orale possono impedire la comunicazione. Sono disponibili dispositivi che consentono la comunicazione utilizzando dei leggeri movimenti della mano o grazie alla tecnologia “eye-gaze” che traccia i movimenti oculari.
Alimentazione I problemi alimentari legati alla masticazione, alla deglutizione e al portare fisicamente il cibo alla bocca possono essere un problema. È necessario garantire un’alimentazione adeguata. Può essere difficile mantenere l’apporto calorico necessario per la salute delle ossa, la crescita e lo sviluppo o evitare la sovralimentazione. Altezza, peso e indice di massa corporea (BMI) devono essere costantemente monitorati. Un dietologo può aiutarvi con una dieta appropriata che funzioni al meglio per il bambino. Un ergoterapista può fornirvi le attrezzature adattive.
Problemi gastrointestinali (GI) Le difficoltà di masticazione, le diete liquide e il movimento limitato possono rallentare il sistema digestivo. Ciò può provocare reflusso acido, sensazione di sazietà dovuta allo svuotamento lento dello stomaco o al rallentamento della funzione intestinale, vomito e stitichezza. Possono verificarsi problemi con il metabolismo, la glicemia, la scomposizione dei grassi e ipertensione. L’esercizio fisico migliora la digestione e il metabolismo degli zuccheri. Anche la “terapia dello standing (stare in piedi)” favorisce la digestione. I gastroenterologi sono specializzati in problemi gastrointestinali. Possono essere necessari farmaci per i problemi di cui sopra.
Ortopedia: i problemi con lo scheletro sono un aspetto preoccupante della SMA. L’ortopedico, un professionista sanitario esperto nella cura delle ossa, può essere utile non solo per trattare i problemi ossei, ma anche per evitarli.
Lo stretching delle articolazioni è importante per mantenerle elastiche. È un’attività che può essere fatta per il bambino se non è in grado di muovere il corpo da solo. Questo permette di evitare contratture e crampi e fornisce un comfort generale al corpo.
Le stecche e i tutori possono essere utilizzati per mantenere il corpo in posizione, stabilizzare le articolazioni e favorire il movimento. Possono essere utilizzati sulle braccia, sulle gambe, sul tronco, sul collo o ovunque sia necessario un posizionamento. Alcuni vengono indossati solo di notte.
Le contratture o l’irrigidimento delle articolazioni sono un problema comune nella SMA, poiché il movimento è messo a dura prova. Il trattamento terapeutico sarà fornito e insegnato al bambino e ai familiari per mantenere o migliorare la flessibilità e rallentare la progressione del deperimento muscolare, oltre a fare movimento ed esercizio fisico. Le contratture possono essere ridotte con un intervento chirurgico ortopedico o una serie di ingessature in cui l’articolazione viene rilasciata nel tempo.
Il posizionamento è importante per il mantenimento del corpo e della sua funzionalità. L’attrezzatura per ottenere l’allineamento anatomico del corpo contribuirà a mantenere il corpo in forma, efficace e funzionale. Possono essere utili esercizi di stretching, stecche e ingessature in serie.
Gli stabilizzatori (standing frame) aiutano il bambino ad assumere una posizione eretta che gli permette di vedere il mondo alla sua altezza naturale, migliora le interazioni personali in alcune situazioni, migliora lo sviluppo osseo e può aiutare la postura e la digestione. Verificare con il medico curante se il bambino è fisicamente pronto per questa attività, per evitare complicazioni inaspettate, soprattutto delle ossa o dei sistemi vascolari.
La scoliosi (spina dorsale con la forma di una “S”) e la cifosi (spina dorsale che si piega in avanti come una “C”) sono preoccupanti a causa della debolezza dei muscoli del tronco. Gli squilibri muscolari possono disallineare le vertebre della colonna vertebrale e ridurre la capacità di respirare bene. Il controllo della scoliosi può essere ottenuto con l’esercizio fisico, i tutori e, se necessario, l’intervento chirurgico. Se è necessario un intervento chirurgico, si può scegliere tra la stabilizzazione rigida della colonna vertebrale e la stabilizzazione flessibile. Le ultime barre di stabilizzazione flessibile consentono un leggero movimento del tronco una volta guariti.
L’allineamento dell’anca è un elemento critico sia quando il bambino è allettato sia quando può stare seduto L’allineamento dei fianchi e l’allungamento dei muscoli dell’anca mantengono il corpo in una buona posizione che evita problemi di contratture dell’anca, disallineamento e scoliosi.
La mobilità e l’esercizio fisico sono fondamentali per tutti. Il corpo brama il movimento. L’esercizio fisico giova a respirazione, digestione, salute mentale e salute in generale. I dispositivi di mobilità come sedie a rotelle, deambulatori, stecche e tutori aiutano il bambino a muoversi nel suo ambiente. Questo crea una sorta di indipendenza nel suo mondo, per quando sarà pronto per evolversi. L’esercizio fisico nel corso della terapia sarà prescritto su base individuale. Altre opzioni concernenti l’esercizio fisico possono includere giochi di movimento, idroterapia, sport adattivi o qualsiasi altra cosa che incoraggi il funzionamento del corpo.
Salute delle ossa Quando il peso corporeo non viene trasferito alle ossa, i minerali tendono a diminuire, rendendo le ossa fragili e quindi facilmente fratturabili. L’osteopenia è una perdita di densità ossea che porta all’osteoporosi. I test di densità ossea dovrebbero essere eseguiti annualmente sui soggetti affetti da SMA di tutte le età, per individuare precocemente problemi con la densità ossea in modo da poter iniziare il trattamento. L’esercizio fisico e la posizione eretta aiutano a mantenere la densità ossea, ma alcuni casi sono più gravi e richiedono l’assunzione di farmaci.
L’equipe per le Cure Palliative è composta da un gruppo di persone, tra cui infermieri appositamente formati, che assisteranno il bambino con un supporto medico per le esigenze relative alla qualità della vita. Questo servizio non si limita alle cure di fine vita, ma garantisce che le esigenze di vostro figlio, e le vostre, siano soddisfatte. La SMA può evolvere in molte direzioni. Questo gruppo può iniziare le cure su vostro figlio o ritirarsi, a seconda delle esigenze che cambiano.
Storia
1891 – La SMA fu descritta per la prima volta nella letteratura medica da Gundo Werdnig quando la condizione fu osservata in due fratelli neonati. Ulteriori descrizioni di tale debolezza progressiva nei neonati vennero descritte nel 1893-1900 da Johan Hoffmann, quando venne coniato il termine di “atrofia muscolare spinale”. Werdnig e Hoffmann diedero il nome alla SMA di tipo I, ma le loro descrizioni riguardavano casi intermedi.
Casi gravi furono presentati nel 1899 e nel 1903. Negli anni Cinquanta sono stati presentati dei casi più lievi di SMA con deambulazione e posizione eretta. Ci sono state controversie sui tipi e sulle classificazioni fino al 1991, quando l’Associazione per la Distrofia Muscolare ha sponsorizzato il sistema di classificazione nel corso di un Consorzio Internazionale sull’Atrofia Muscolare Spinale basato su tre tipi di SMA e sull’età di insorgenza.
Solo nel 1995 il laboratorio Melki ha stabilito che la SMA è causata da una delezione nel gene SMN1 sul cromosoma 5Q13 e SMN2. È stata una scoperta cruciale, poiché prima di essa non si conosceva nessuna ragione specifica per lo sviluppo della SMA e quindi l’evoluzione di un trattamento non era determinabile. Entro cinque anni dalla scoperta genetica, è stato possibile creare dei modelli animali per studiare la SMA e iniziare a svilupparne i trattamenti.
Nel 2007, Wang e colleghi hanno sviluppato la prima linea guida clinica per la cura delle persone affette da SMA, compreso l’invecchiamento con la SMA.
Da quando sono stati compresi i problemi genetici, l’Istituto Nazionale per i Disturbi Neurologici e l’Ictus (National Institute of Neurological Disorders and Stroke), le fondazioni, i farmacologi e i biotecnologi hanno sviluppato terapie per aumentare o migliorare SMN1 e SMN2 utilizzando terapie con piccole molecole, terapie con RNA e terapie geniche. I risultati sono stati significativi e sono disponibili i seguenti trattamenti terapeutici:
- 2016 Nusinersen (SPINRAZA) è stato approvato dall’FDA per tutti i tipi di SMA.
- 2017 Onasemnogene abeparvovec-xioi (Zolgensma) è stato approvato dall’FDA per la sostituzione del gene SMN1.
- 2020 Risdiplam (Evrysdi) è stato approvato dall’FDA per stimolare l’espressione del gene SMN2 della proteina SMN. Si tratta del primo farmaco orale per il trattamento della SMA.
La ricerca è aumentata costantemente, con un numero sempre maggiore di studi condotti e in vari stati di sperimentazione clinica. Inoltre, è importante ricordare che molti altri ricercatori hanno contribuito a migliorare la vita delle persone affette da SMA, per mezzo della loro ricerca sui miglioramenti della respirazione, sui farmaci, sul trattamento della scoliosi e di altri problemi ossei, oltre che all’attenzione per la qualità della vita.
Fatti e cifre
La SMA è la causa più comune di morte dovuta alle alterazioni genetiche nei neonati.
Un bambino su 6.000 nasce con la SMA. Un individuo su 11.000 di tutte le età è affetto da SMA.
Coloro che sono portatori dell’anomalia genetica della SMA, ma che non presentano la condizione, sono 1 su 54.
Il tipo di SMA più comune è la malattia di Werdnig-Hoffmann, responsabile dell’80% dei casi. La malattia di Kugelberg-Welander è un altro tipo che viene diagnosticato nella prima infanzia.
Lo sviluppo della SMA nell’infanzia e nella prima giovinezza è associato a esiti più sfavorevoli rispetto a quando si sviluppa più tardi nella vita.
Risorse
Per ulteriori informazioni sull’atrofia muscolare spinale o per domande specifiche, i nostri consulenti specializzati (Information Specialist) sono disponibili dal lunedì al venerdì al numero verde 800-539-7309.
